Integrated transcriptome landscape of ALS identifies genome instability linked to TDP-43 pathology
Journal club:
Integrated transcriptome landscape of ALS identifies genome instability linked to TDP-43 pathology
Why read this paper?
Focuse on ALS research using the same key technologies: human induced pluripotent stem cell-derived motor neurons (iPSMNs) and RNA sequencing.
Which journal?
Nature communication (Published: 20 April 2023)
Who wrote the paper?
Dr Oliver J. Ziff: The Francis Crick Institute, London. Research focus on investigating the causes of Amyotrophic Lateral Sclerosis (ALS) using bioinformatic multi-omic approaches.
Professor Rickie Patani: University College London, London. A physician scientist with over a decade of direct experience working on human induced pluripotent stem cell (iPSMNs) models of neurodegeneration.
What is ALS
- Amyotrophic lateral sclerosis (ALS), is a nervous system disease that affects nerve cells in the brain and spinal cord. ALS causes loss of muscle control. ALS often begins with muscle twitching and weakness in an arm or leg, trouble swallowing or slurred speech. Eventually ALS affects chewing, swallowing, speaking and breathing. The disease gets worse over time.
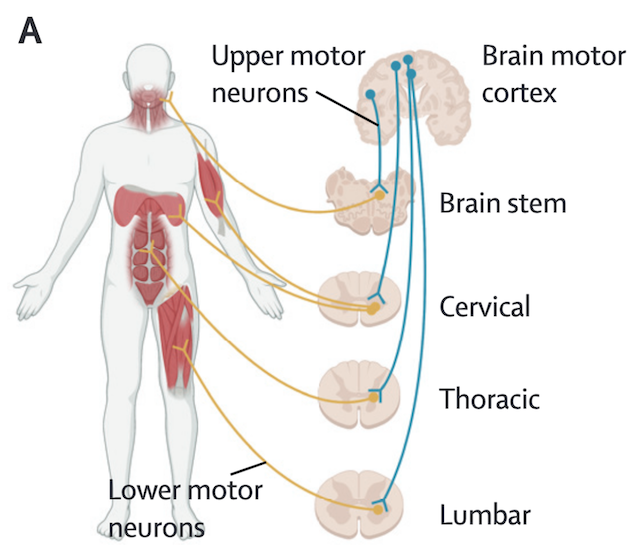
- ALS cases can be grouped by two categories:
- About 10% of ALS cases are inherited and referred to as familial ALS. More than 20 gene mutations have been established to cause ALS, the most common being is C9orf72, SOD1, TARDBP(The gene encoding TDP-43) and FUS.
- Sporadic ALS technically refers to cases without a family history, the term is sometimes also used to describe cases without a identifiable genetic basis.
Background
- A major hurdle in identifying the causes of ALS is the inaccessibility of patient motor neuron. Post-mortem tissue has been used to study ALS.
- TDP-43 proteinopathy remains a post-mortem pathologic diagnosis of ALS, which is observed in 97% of cases and is characterised by the mislocalisation and aggregation of TDP-43 in the cytoplasm of neurons.
- Post-mortem tissue represents the end stages of the disease and contains very few surviving motor neurons. Human induced pluripotent stem cell-derived motor neurons (iPSMNs) allow researchers to model ALS motor neuron degeneration at earlier stages and enable sporadic ALS to be modelled. The ability to differentiate patient-derived induced pluripotent stem cells (iPSCs) into motor neurons (iPSMNs) has transformed ALS modeling.
Study Objective
This research uses data from ALS iPSMNs and post-mortem tissue to perform an integrated transcriptome analysis.
The goal is to understand how various genetic backgrounds contribute to ALS and to map the landscape of RNA changes related to the disease.
Cohort Design
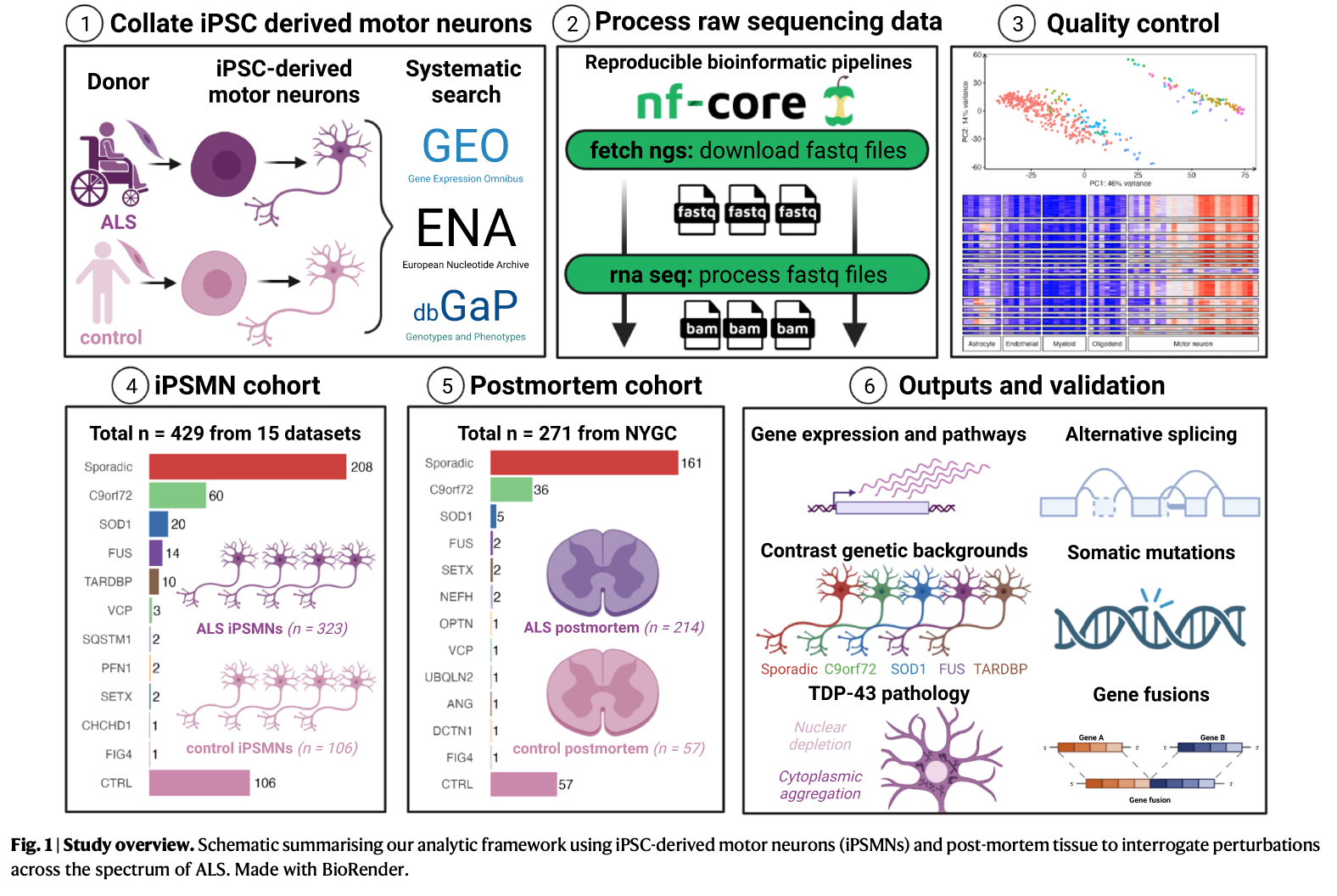
iPSMN cohort:
Online database search (from GEO,SRA, EBI arrayExpress, ENA, synapse.org, etc.) identified 16 ALS iPSMN bulk RNA-sequencing (RNA-seq) datasets, which undergone short-read bulk RNA-sequencing (RNA-seq) from human iPSMN samples derived from individuals with ALS and non-ALS controls.
After quality control, 15 dataset remained:
In total 429 iPSMNs, of which 323 were from ALS patients and 106 were from non-ALS controls.
Quality control (Excluded datasets that:)
(1) had not undergone an accepted spinal motor neuron differentiation protocol using the steps detailed in Sances et al.
(2) failed RNA-seq quality control measures
(3) inadequate neuronal marker expression
(4) exhibited unadjustable batch effects between ALS and control samples (e.g. different RNA library strategies or sequencing platforms, such as ALS (Ribo-Zero) and control (polyA) samples).
Samples clustered into two groups based on library preparation (poly-adenylated or total ribosomal depletion).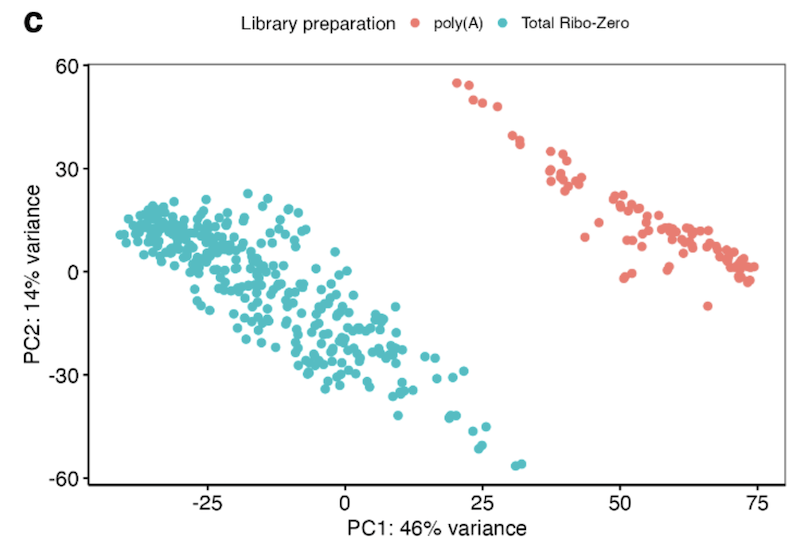
Scatterplot of PC1 against PC2 individual gene loadings of the top 500 most variable genes: Principal component gene loadings confirmed that the separation was driven by histone and small nucleolar encoding genes, which represent non-polyadenylated genes.
(Loading plot can be used to identify which variables have the strongest influence on each component. The coefficients can range from -1 to 1. Coefficients close to -1 or 1 indicate that the variable has a strong influence on the component.)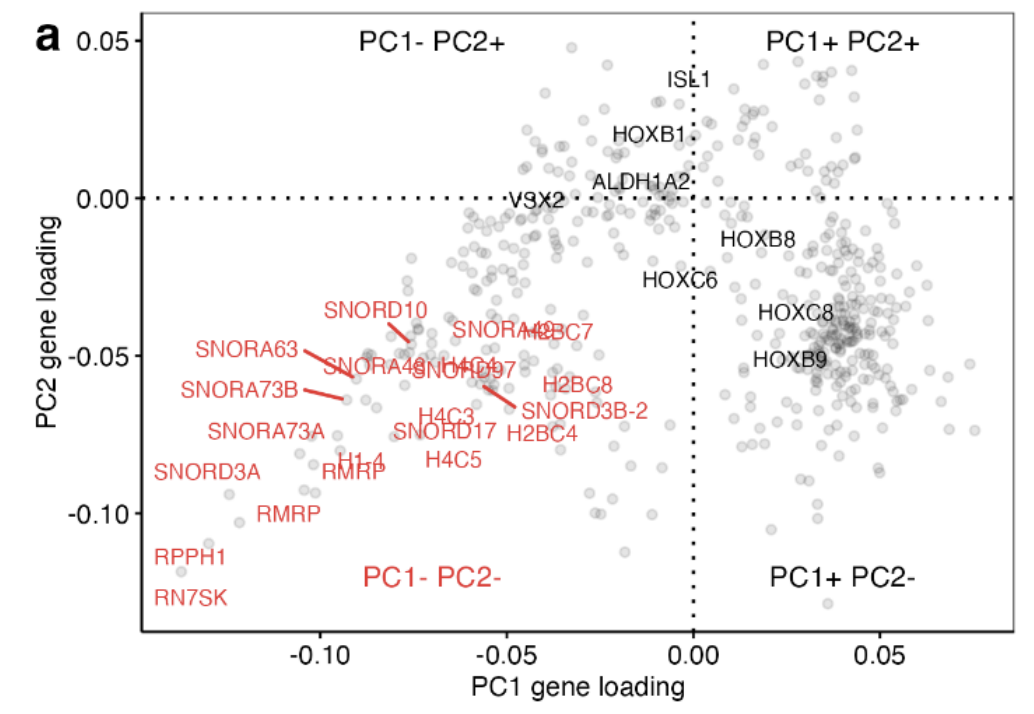
ALS iPSMNs carried pathogenic mutations in 10 different genes, including C9orf72 (n = 60), SOD1 (n = 20), FUS(n = 14), TARDBP (n = 10), VCP (n = 3) and other ALS mutations (n = 9)
Sporadic ALS(patients without an identifiable ALS mutation): 208 (64.2%)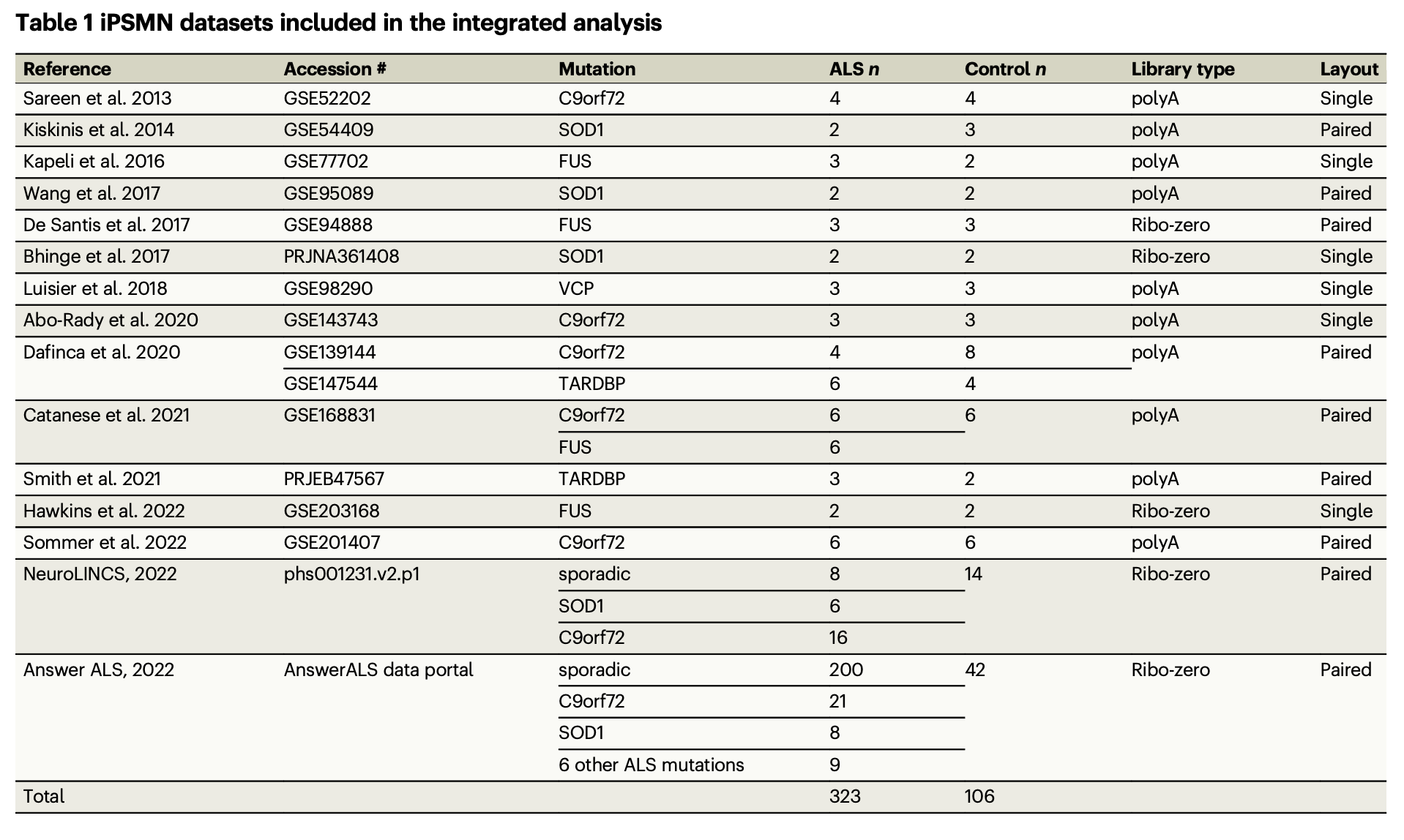
Postmotrem cohort (NYGC cohort)
Post-mortem spinal cord ALS RNA-seq samples were derived from samples from the New York Genome Centre (NYGC) ALS consortium, consisting of tissue from 214 ALS patients and 57 controls.
Samples from non-spinal cord sites were excluded.
Post-mortem subgroup analyses were limited by the sample size for FUS (n = 2) and there were no TARDBP mutants available.
TDP-43 depletion and overexpression
- The post-mortem brain FACS sorted neuronal nuclei depleted of TDP-43 dataset was acquired from accession GSE126543. Fluorescence-activated cell sorting (FACS) was used to enrich neuronal nuclei with and without TDP-43 from postmortem brain tissue from patients with frontotemporal dementia and ALS (FTD–ALS). Differential expression results were calculated by comparing TDP-43 negative versus TDP-43 positive samples.
- TDP-43 knockdown datasets: 7 dataset combination
- Primary mouse neurons overexpressing TDP-43: GSE162048
Results
ALS iPSMNs activate the DNA damage response(Pan-ALS gene expression changes)
All library preparation together:
Differential expression gene (DEG)
DESeq2
323 ALS vs 106 control iPSMNs
The Wald test, controlling for sex differences and dataset variation with the design formula ∼ sex + dataset + condition.
Upregulated: 20
Downregulated: 23
Amongst differentially expressed genes most increased in ALS was the endoribonuclease RNase L (RNASEL) which regulates the decay of cytoplasmic RNA and localisation of RNA binding proteins (RBPs)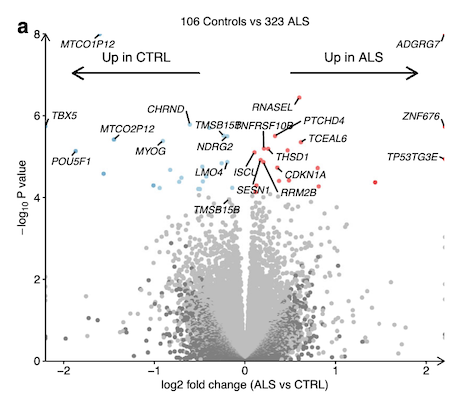
Functional over-representation analysis (ORA)
Upregulated:
the DNA damage response (hypergeometric FDR = 2.2 × 10−5; SESN1, RRM2B, TNFRSF10B)
p53 signalling (FDR = 2.7 × 10−5; CDKN1A, TP53TG3E, FBXO22)
Downregulated:
DNA-binding transcription factor activity (FDR = 0.003; MYOG, TBX5, POU5F1)
ventral spinal cord development (FDR = 0.004; LMO4, OLIG2, FOXN4; Fig. 2b).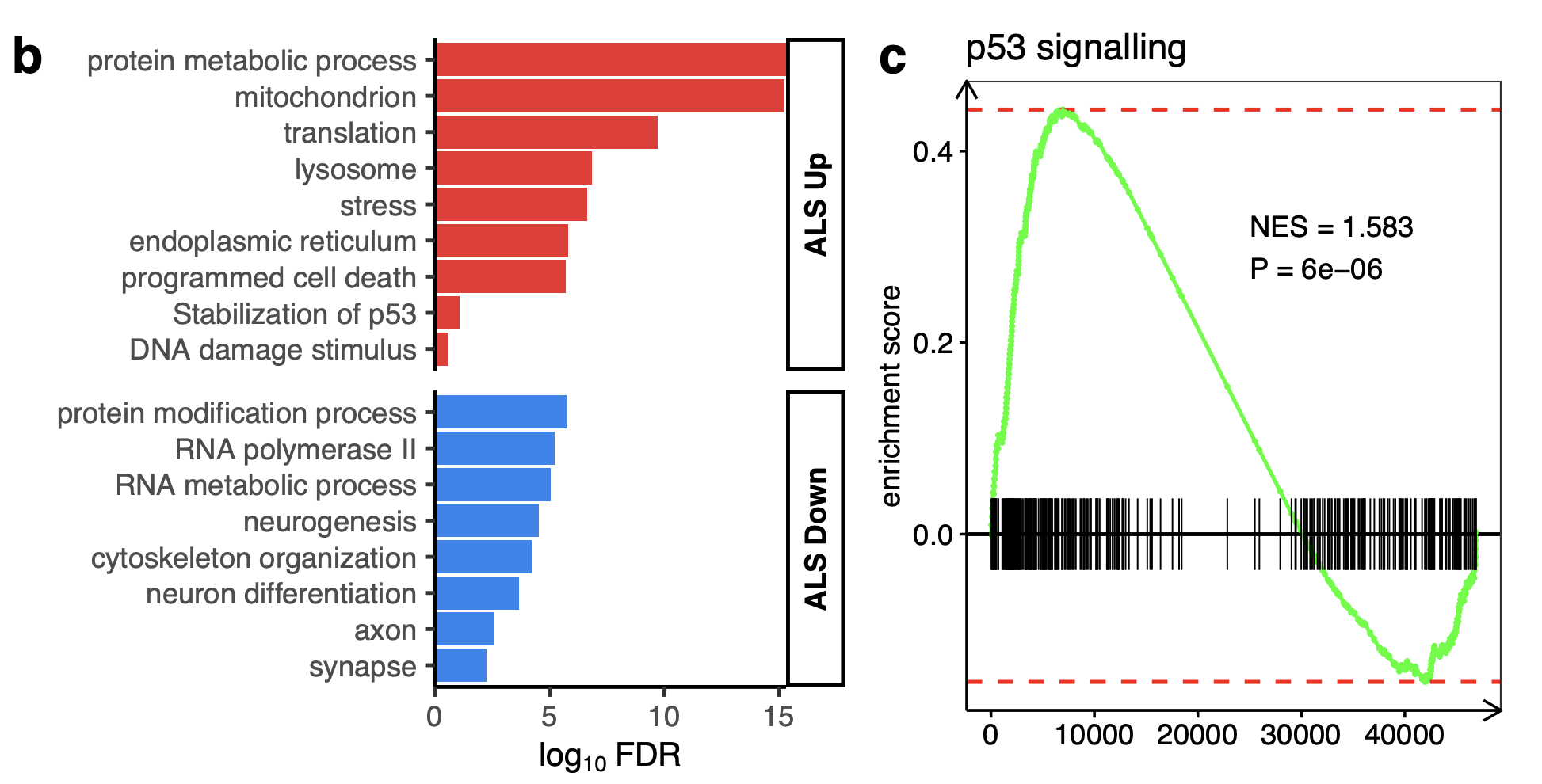
Gene Set Enrichment Analysis (GSEA)
Upregulated:
p53 signal transduction gene set (GO:0072331, n = 264)
The mitotic G1 DNA damage checkpoint (daughter pathway of DNA damage response)
Intrinsic apoptosis signalling (daughter pathway of intrinsic apoptotic signaling pathway by p53 class mediator).
Signalling Pathway RespOnsive GENes (PROGENy) analysis(Ref 2)
PROGENy is a method developed to infer pathway activity from gene expression data. The scores are calculated using a linear models with weights based on a common core of pathway responsive genes (consensus gene signatures) obtained from publicly available perturbation experiments.
Inside PROGENy, one can find gene signatures for 14 different pathways: Androgen,Estrogen,EGFR,Hypoxia,JAK-STAT,MAPK,NFkB,PI3K,p53,TGFb,TNFa,Trail,VEGF and WNT
Upregulated:
p53 (NES + 13.0, p < 0.001): Examining each gene in the p53 pathway according to its p53 weighting in PROGENy revealed that the genes with the strongest responsiveness in p53 activity in ALS iPSMNs included CDKN1A, SESN1, RRM2B, MDM2, C2orf66, ZNF561 and ZMAT3 (Fig. 2e).
Mitogen-Activated Protein Kinase (MAPK; NES + 5.6, p < 0.001)
Downregulated:
WNT (NES −2.5, p = 0.03; Fig. 2d)
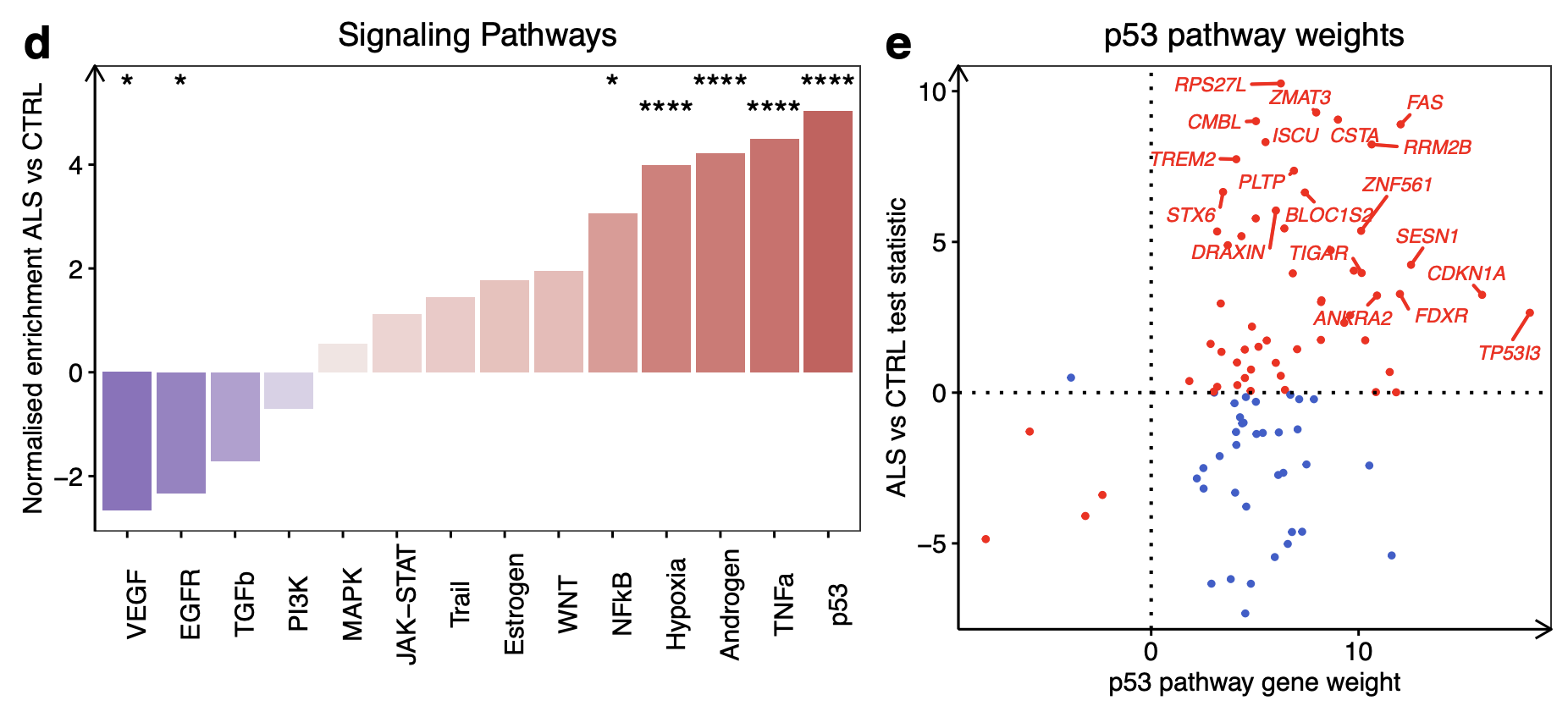
e Expression changes of p53 signalling pathway genes in ALS versus control according to their PROGENy weights. Genes increasing p53 activity in ALS are red whilst genes decreasing p53 activity in ALS are blue.
Transcription factors activity (within the DoRothEA database)
The activities of 429 transcription factors (TFs) from their regulon expression within the DoRothEA database.
Upregulated:
TP53 was the TF with the greatest increase in activity in ALS (NES + 7.62, p < 0.001)
ZNF274
ATF4
Downregulated:
PRDM14
ZNF263
SIX5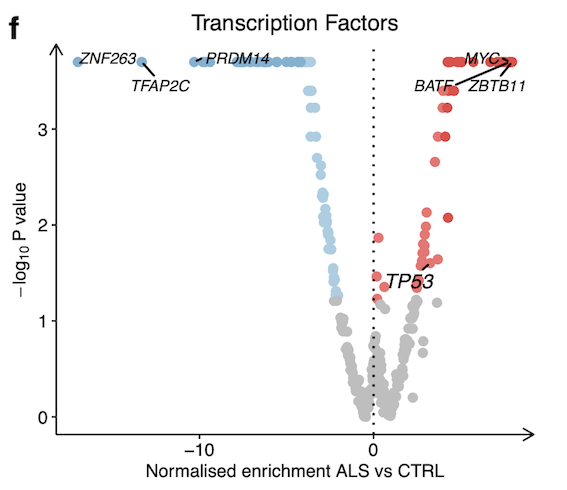
subgroup analysis in library preparation separately
subgroup analysis in poly(A) datasets and total Ribo-Zero separately:
Poly(A) datasets: (10 datasets; 48 ALS, 43 control iPSMNs)
Total Ribo-Zero (5 datasets; 275 ALS, 63 control iPSMNs)
Differential expression gene (DEG): poly(A) datasets there were 69, and in total Ribo-Zero datasets there were 12
RNase L was significantly increased in ALS iPSMNs in both library preparation analyses independently.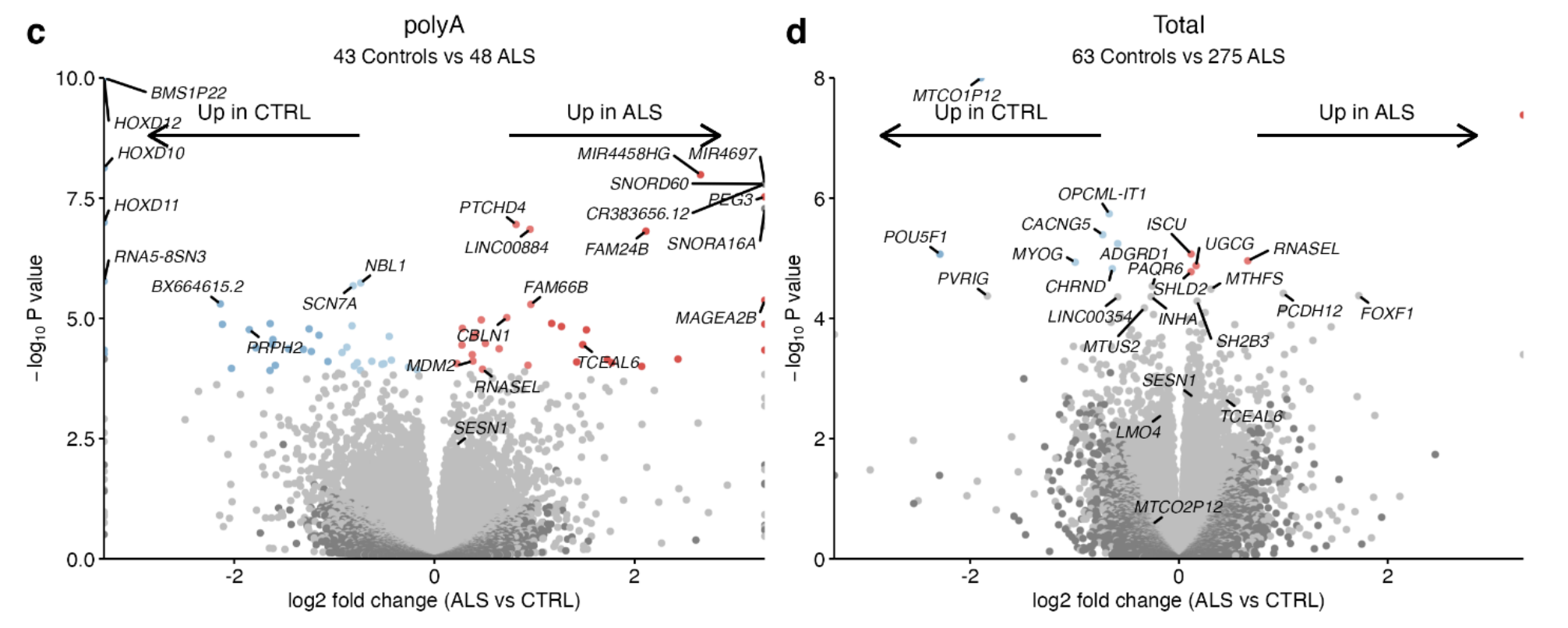
PROGENy: significant increases in p53 pathway activity in ALS with both library preparations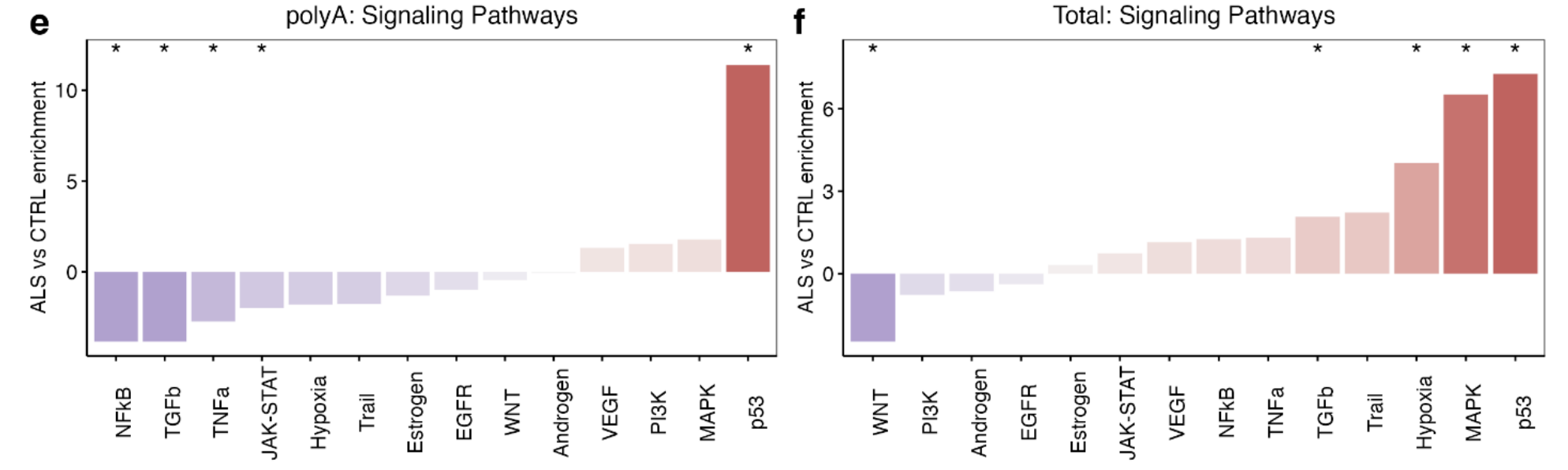
Activity of transcription factors within the DoRothEA database:
TP53 TF activity was significantly increased in ALS iPSMNs in both library preparation groups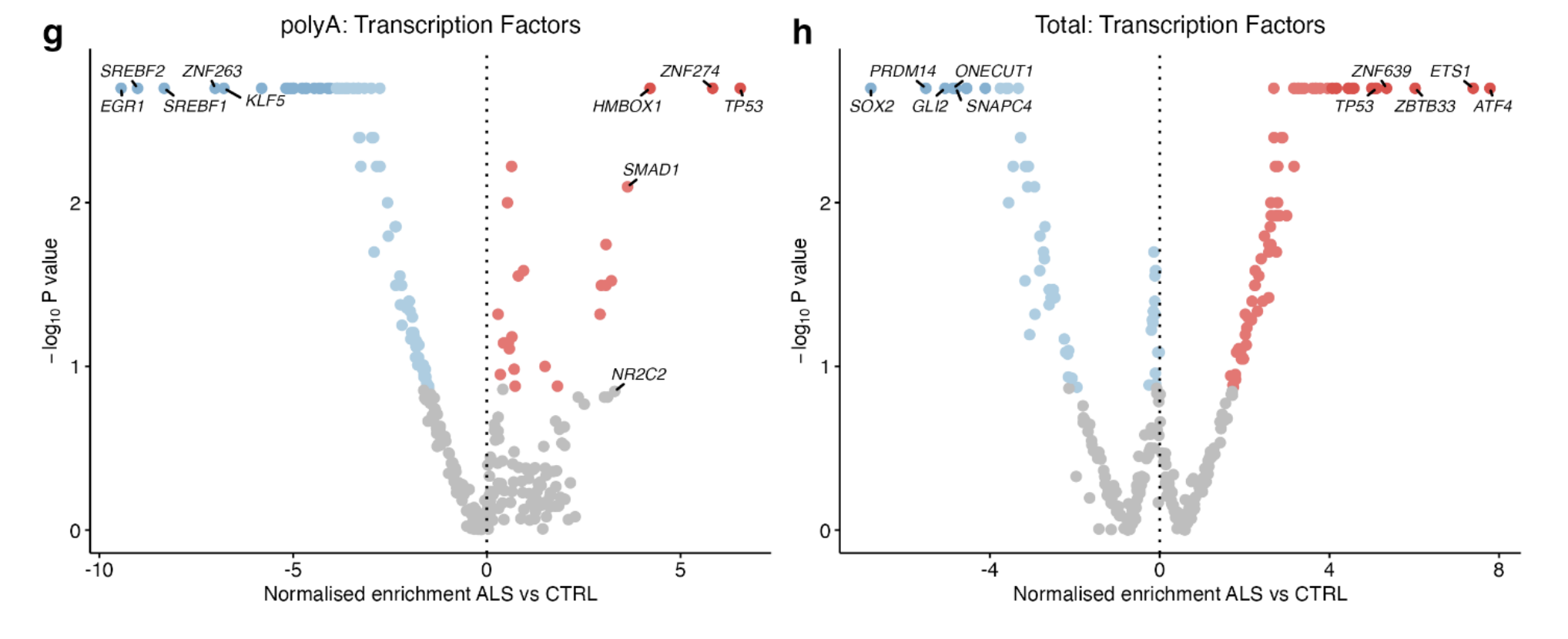
subgroup analysis in dataset separately
Both TP53 transcription factor and p53 signalling were independently upregulated in ALS iPSMNs in 11 of 17 datasets (seems a mistake? why 17 datasets).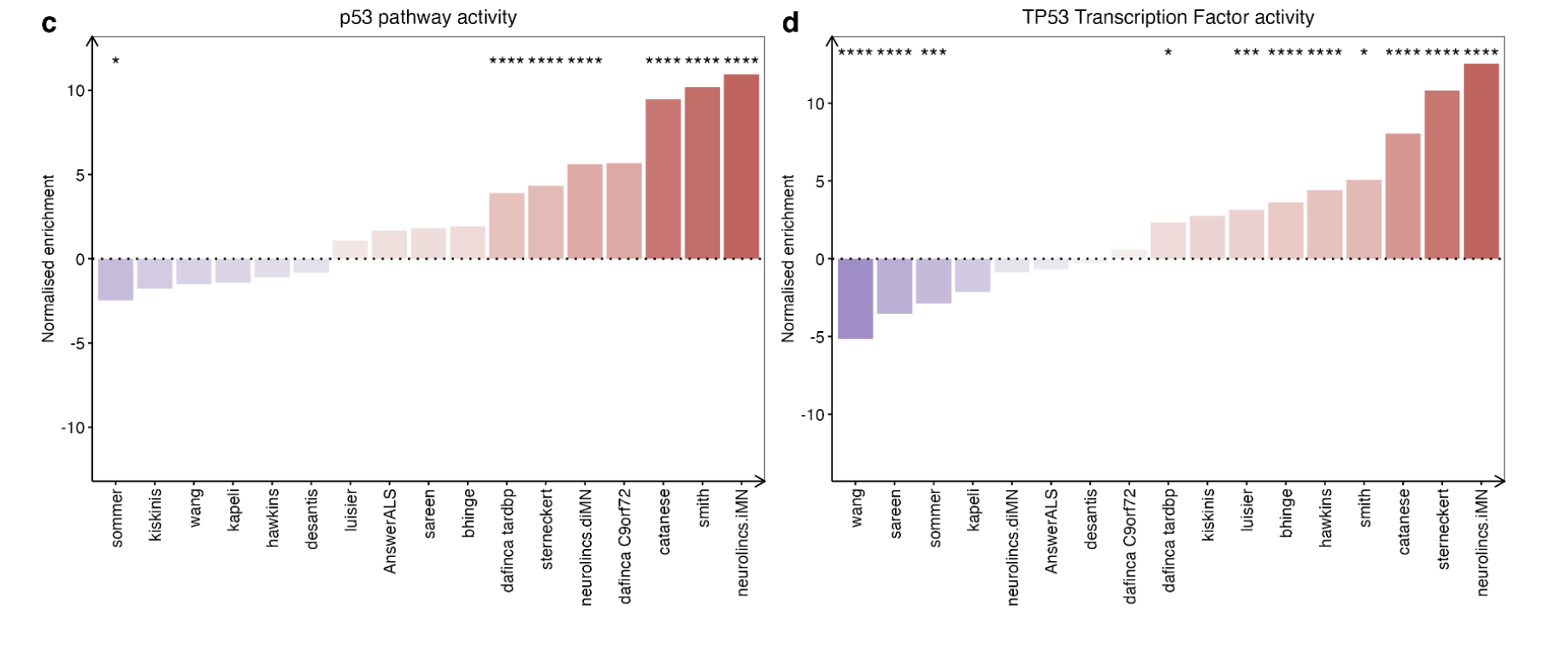
Proteome in Answer ALS dataset
DEP:
no proteins were significant at FDR < 0.05 in ALS compared to control
276 were significantly different at unadjusted p < 0.05 (including p53 pathway components RBBP7, CSNK2B, PRMT1, CNOT9)
ORA:
Upregulated:
protein metabolism (e.g. PSDMD9, NAE1, PSMB5)
RNA metabolism (e.g. APP, CSTF1, CIRBP, SNRPD2)
protein binding (e.g. RRBP1, RPS29, EIF1)
other processes established in ALS pathophysiology (stress response, cholesterol synthesis, nucleocytoplasmic transport)
Downregulated:
Golgi transport
GSEA
p53 pathway nonsignificant increase (NES + 0.98, p = 0.5)
Correlation between mRNA and protein level
Comparing changes in mRNA expression with protein expression revealed a weak inverse correlation, consistent with previous reports showing poor correlations between mRNA and protein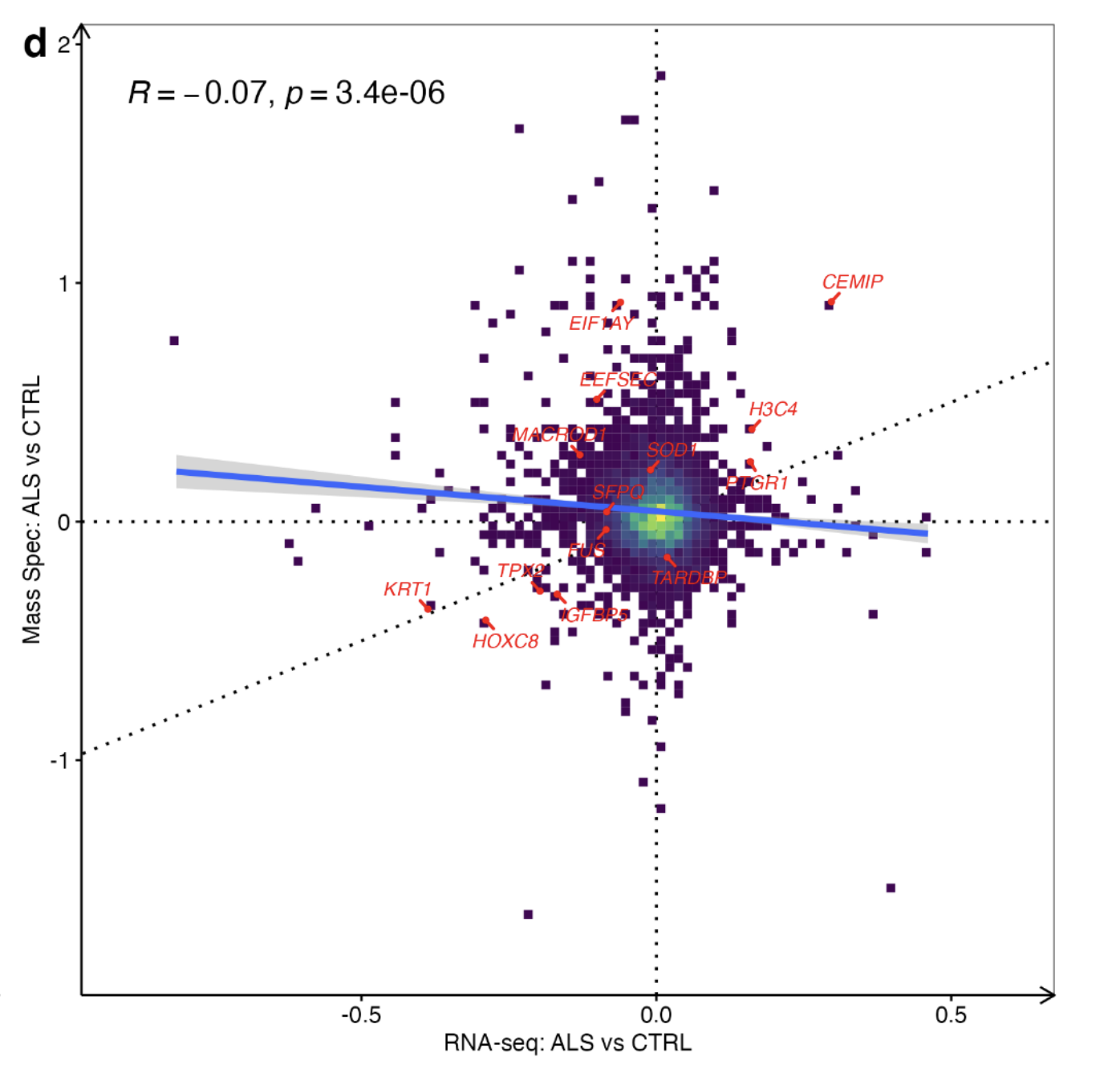
p53 activation across different ALS genetic backgrounds
Of the 15 datasets,
7 included C9orf72 mutants (comprising 60 C9orf72 iPSMNs and 83 control iPSMNs),
5 included SOD1 mutants (20 SOD1, 63 controls),
5 included FUS mutants (14 FUS, 55 controls),
3 included TARDBP mutants (10 TARDBP, 48 controls) and
2 included sporadic iPSMNs (208 sporadic, 56 controls).
Controls from each dataset were utilised only if the dataset had samples from the relevant genetic background.
DEG:
TARDBP mutants (3,547), FUS (239), C9orf72 (161), and SOD1 (7), sporadic ALS(4)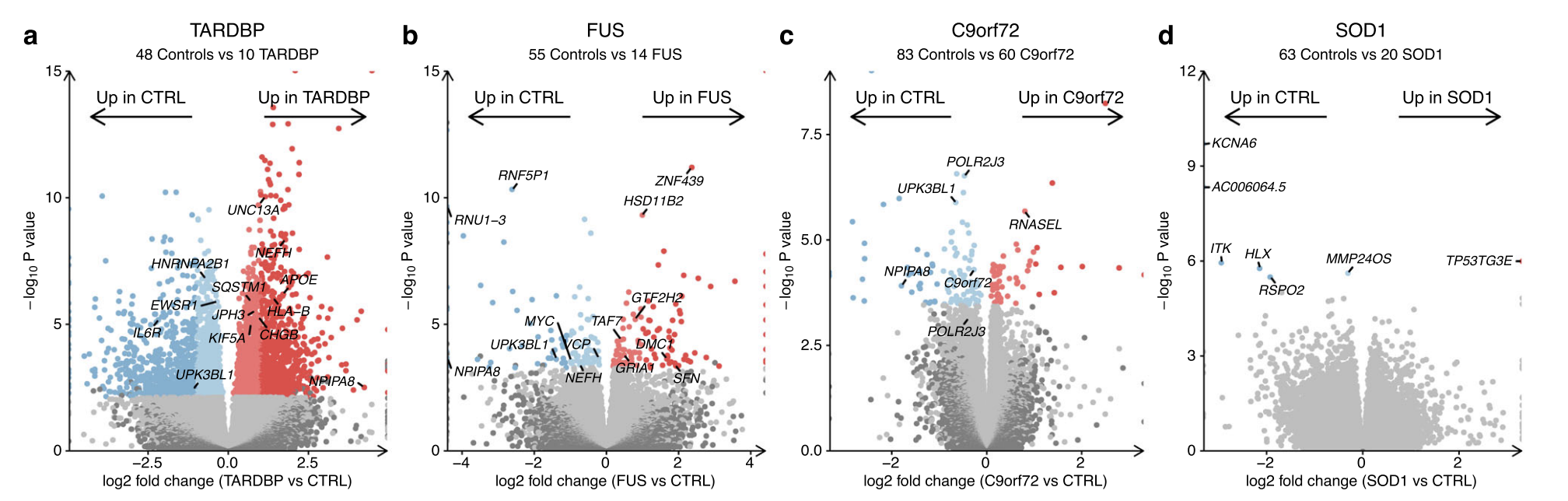
Although no genes were significantly changed in expression across all ALS genetic backgrounds, Uroplakin UPK3BL1 and nuclear pore complex interacting protein NPIPA8 were changed in the C9orf72, TARDBP and FUS subgroups (Supplementary Fig. 12b).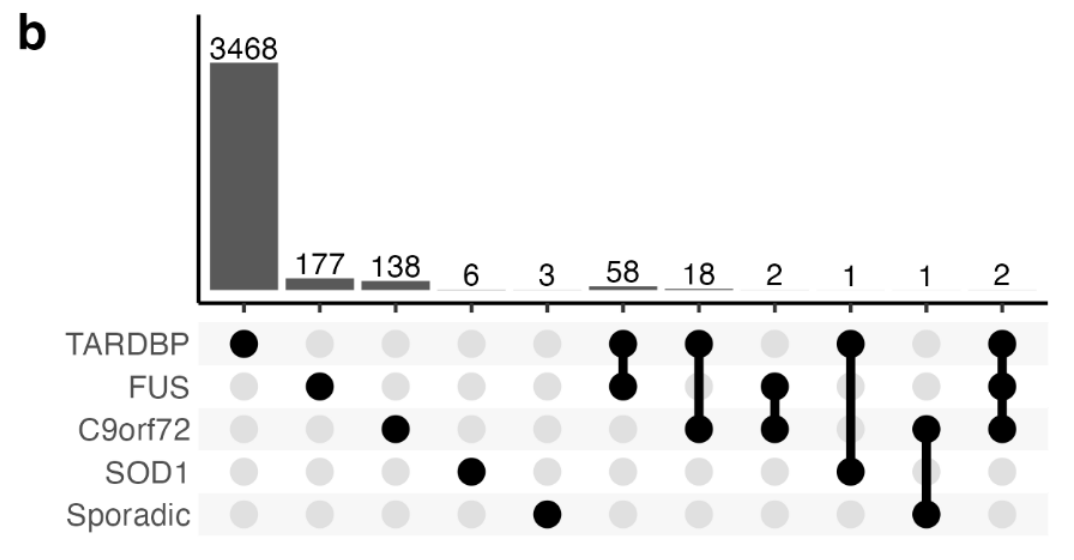
Pearson correlation between ALS genetic backgrounds
Correlating transcriptome-wide gene expression changes between ALS genetic backgrounds revealed weak associations, with the strongest correlation between SOD1 and sporadic lines (Pearson R = + 0.38, p < 2.2 × 10−16) and the weakest between SOD1 and TARDBP (R = −0.14, p < 2.2 × 10−16; Fig. 3f).
ORA
Functional enrichment terms enriched in (c) C9orf72, (d) FUS and (e) TARDBP using the hypergeometric test.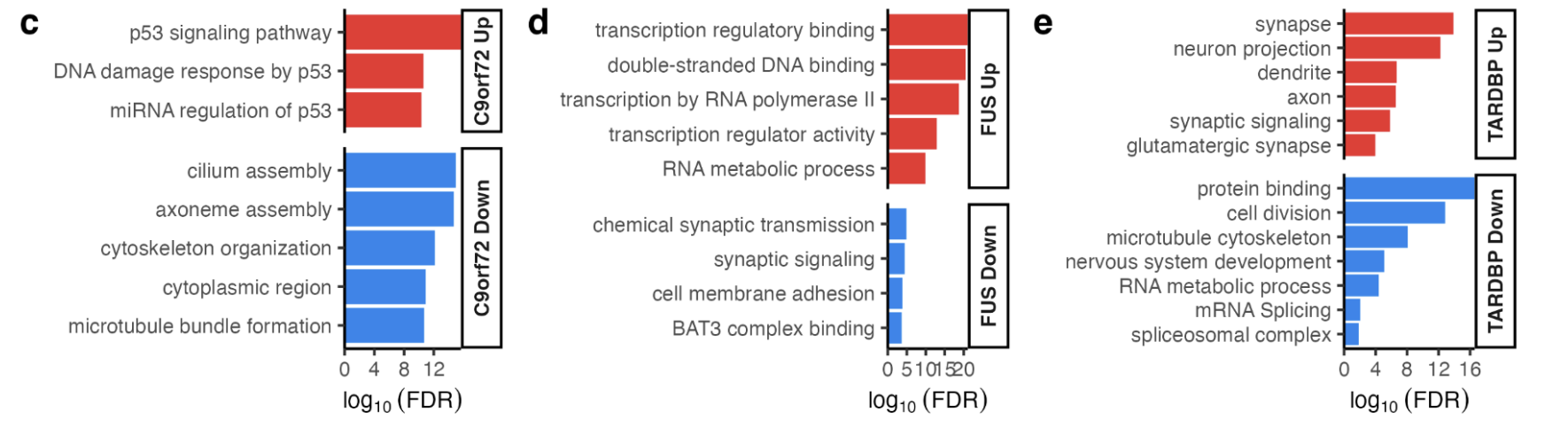
C9orf72 mutants: upregulated genes involved with p53 (hypergeometric FDR = 0.003) and the DNA damage response (FDR = 0.02) whilst downregulating cytoskeleton (FDR = 0.01) and microtubule genes (FDR = 0.02).
FUS mutants: upregulation of genes involved with transcription (FDR = 1.5 × 10−8) and DNA-binding (FDR = 2.4 × 10−8) and downregulation of synaptic signalling genes (FDR = 0.02).
TARDBP mutants: upregulated neuronal (FDR = 7.8 × 10−29) and synaptic genes (FDR = 1.2 × 10−32) and downregulated genes involved in the cell cycle (FDR = 3.1 × 1030) and RNA splicing (FDR = 2 × 10−5, Supplementary Fig. 12c-e).
There were no functional terms enriched amongst SOD1 or sporadic ALS differentially expressed genes.
PROGENy pathway activities
Apart from SOD1 (NES + 0.33, p = 0.16), the p53 pathway activity was significantly increased in each of C9orf72 (NES + 10.9, p < 0.001), TARDBP (NES + 8.6, p < 0.001), sporadic (NES + 4.2, p < 0.001) and FUS (NES + 3.2, p = 0.018; Fig. 3g).
Examining the other signalling pathways revealed that hypoxia, VEGF and MAPK were also increased across most genetic backgrounds, whilst WNT and PI3K tended to be decreased.
TF regulon activity
Significantly increased TP53 activity in C9orf72 (NES + 9.3, p < 0.001), FUS (NES + 4.0, p = 0.004) and TARDBP (NES + 2.5, p = 0.01) but decreased activity in SOD1 (NES −3.0, p < 0.001) whilst sporadic (NES −0.31, p = 0.25) was non-significantly changed.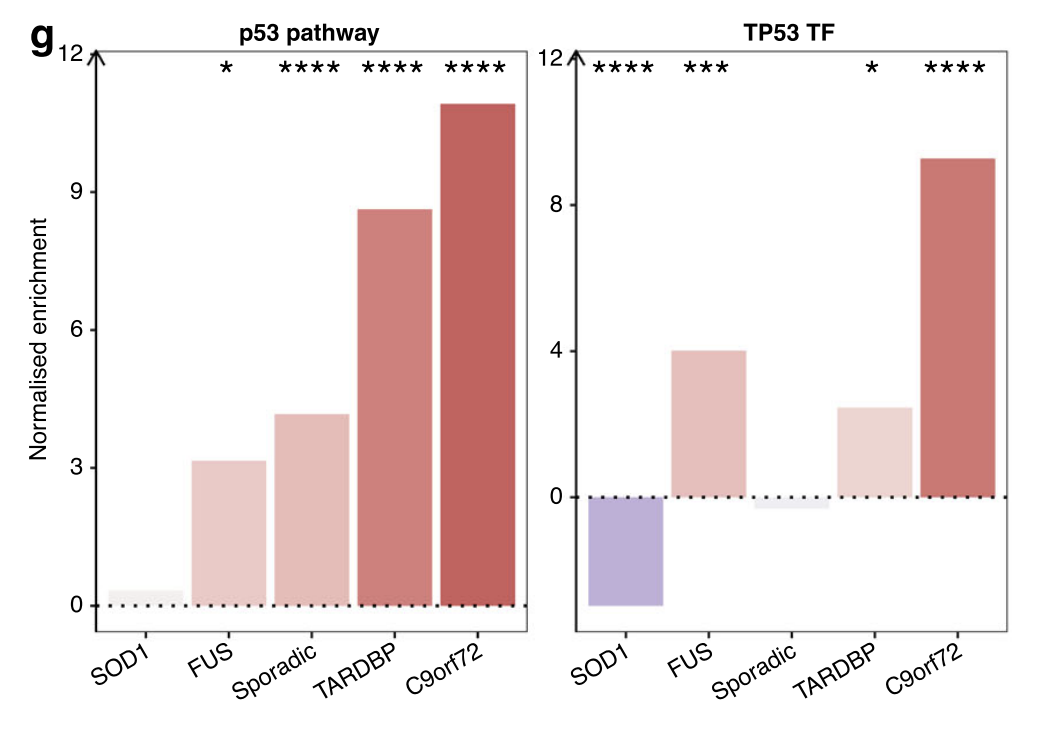
ALS post-mortem tissue shows p53 activation
214 ALS patients and 57 controls
Pan-ALS
Upregulated DEG:
6575 (includes CHIT1, GPNMB and LYZ)
Downregulated:
7489
PROGENy: As with iPSMNs, in ALS post-mortem spinal cord, the most strongly upregulated PROGENy signalling pathway was p53 (NES + 5.0, p < 0.001; Fig. 4d). The top individual genes driving p53 pathway activity in ALS post-mortem included FAS, RRM2B, CSTA, ZMAT3, TP53I3, and CDKN1A ( Fig. 4e)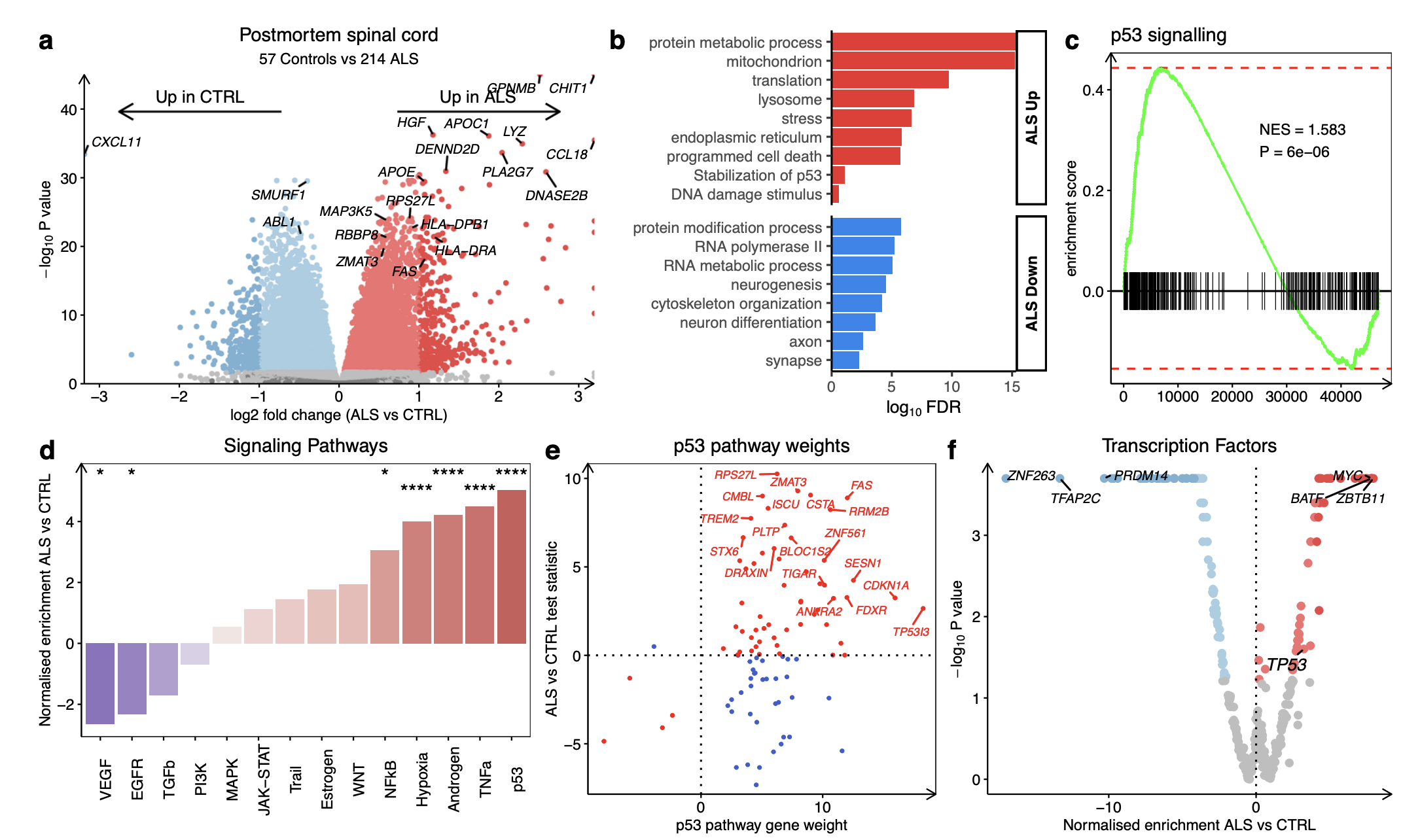
Correlating transcriptome-wide ALS gene expression changes between iPSMNs and post-mortem spinal cord revealed a weak positive correlation (Pearson R = + 0.12, p < 2.2 × 10−16; Fig. 4g).
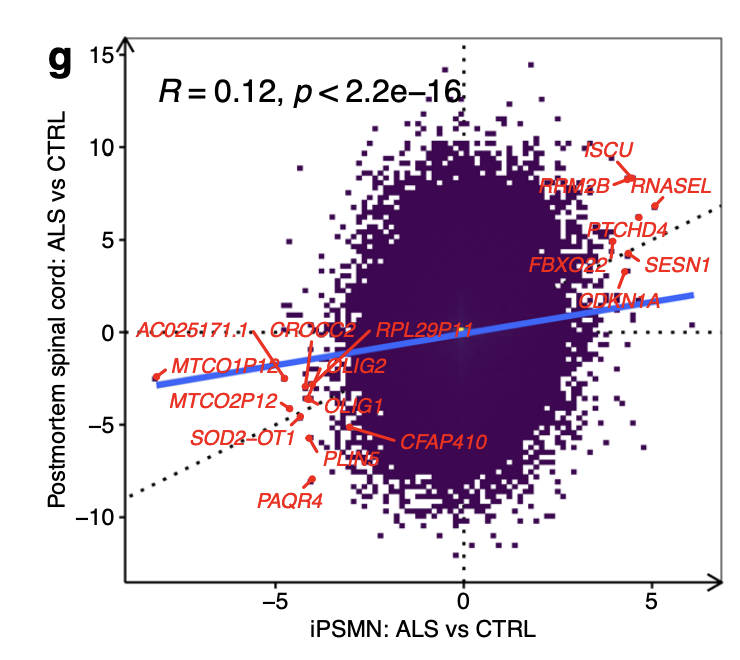
These results not only confirm p53-dependent DNA damage response upregulation in ALS but the overlap between iPSMNs and post-mortem provides insight into motor neuron-specific changes that begin early and persist into the later stages of the disease.
Different ALS genetic backgrounds
In contrast to iPSMNs, comparing genetic subgroups from postmortem spinal cord tissue revealed strongly correlated gene expression changes (R range +0.68 to +0.9) with 1750 overlapping differentially expressed genes between sporadic, C9orf72, SOD1, and FUS subgroups.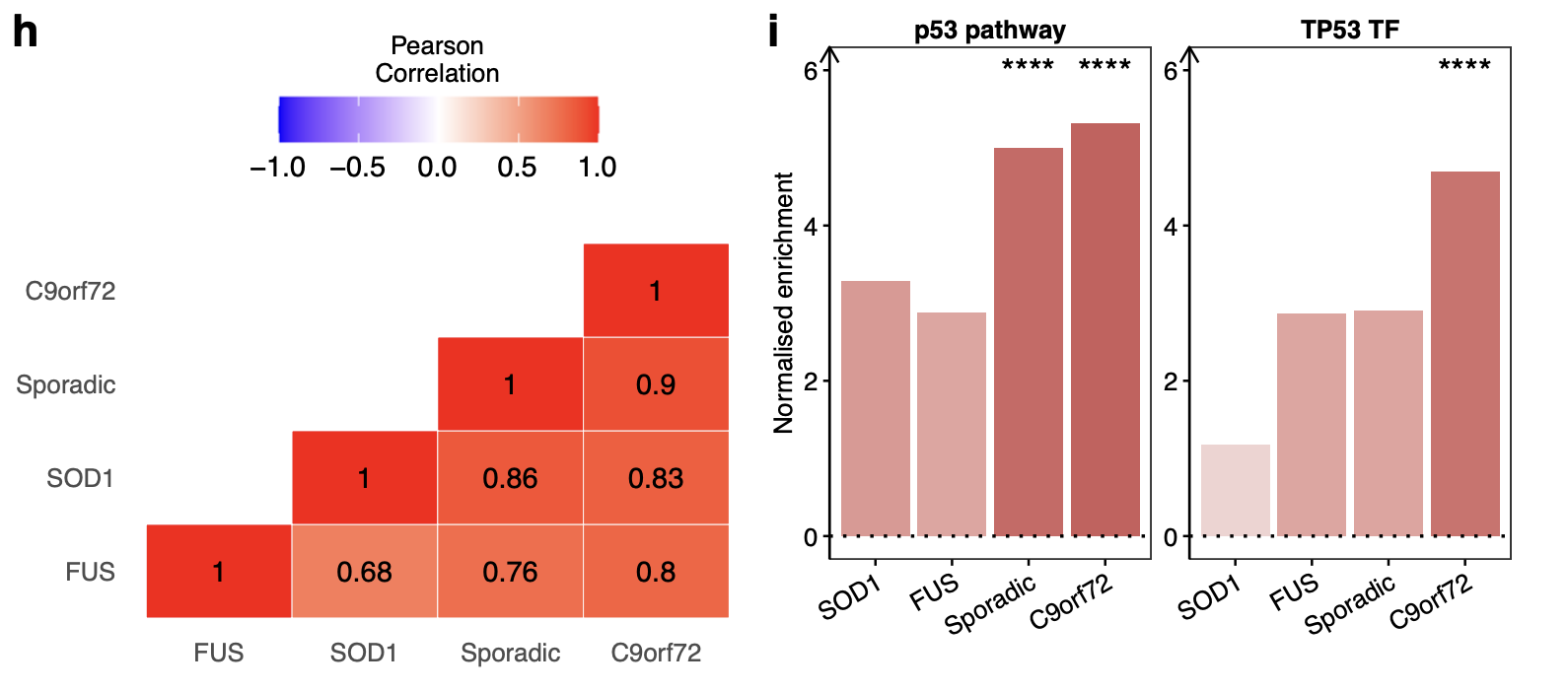
TDP-43 pathology contributes to the DNA damage response
TDP-43, encoded by the TARDBP gene, is an abundant, ubiquitously expressed RNA-binding protein that normally localizes to the nucleus. It has a role in fundamental RNA-processing activities, including RNA transcription, alternative splicing and RNA transport.
A major splicing regulatory function of TDP-43 is to repress the inclusion of cryptic exons during splicing. Unlike normal conserved exons, these cryptic exons occur in introns and are normally excluded from mature mRNAs. When TDP-43 is depleted from cells, these cryptic exons get spliced into messenger RNAs, often introducing frame shifts and premature termination, or even reduced RNA stability.(Ref 3)
Classified ALS samples based on whether their genetic background is associated with TDP-43 pathology:
TDP-43 ALS:
non-TDP-43 ALS: SOD1 and FUS mutant
The findings suggest that the p53 signature from the pan-ALS analyses is largely driven by genetic backgrounds associated with TDP-43 proteinopathy.
To discover whether p53 signalling changes are regulated by TDP-43:
- FACS-sorted neuronal nuclei: with and without TDP-43 pathology from Frontotemporal dementia -ALS (FTD-ALS) post-mortem brain tissue. (Differential expression results were calculated by comparing TDP-43 negative vs TDP-43 positive samples.)
- TDP-43 knockdown cells
- TDP-43 over-expression
Together, these results indicate that both depletion and accumulation of TDP-43 augment p53 activity, suggesting that tight regulation of TDP-43 levels is required to ensure an appropriate DNA damage response.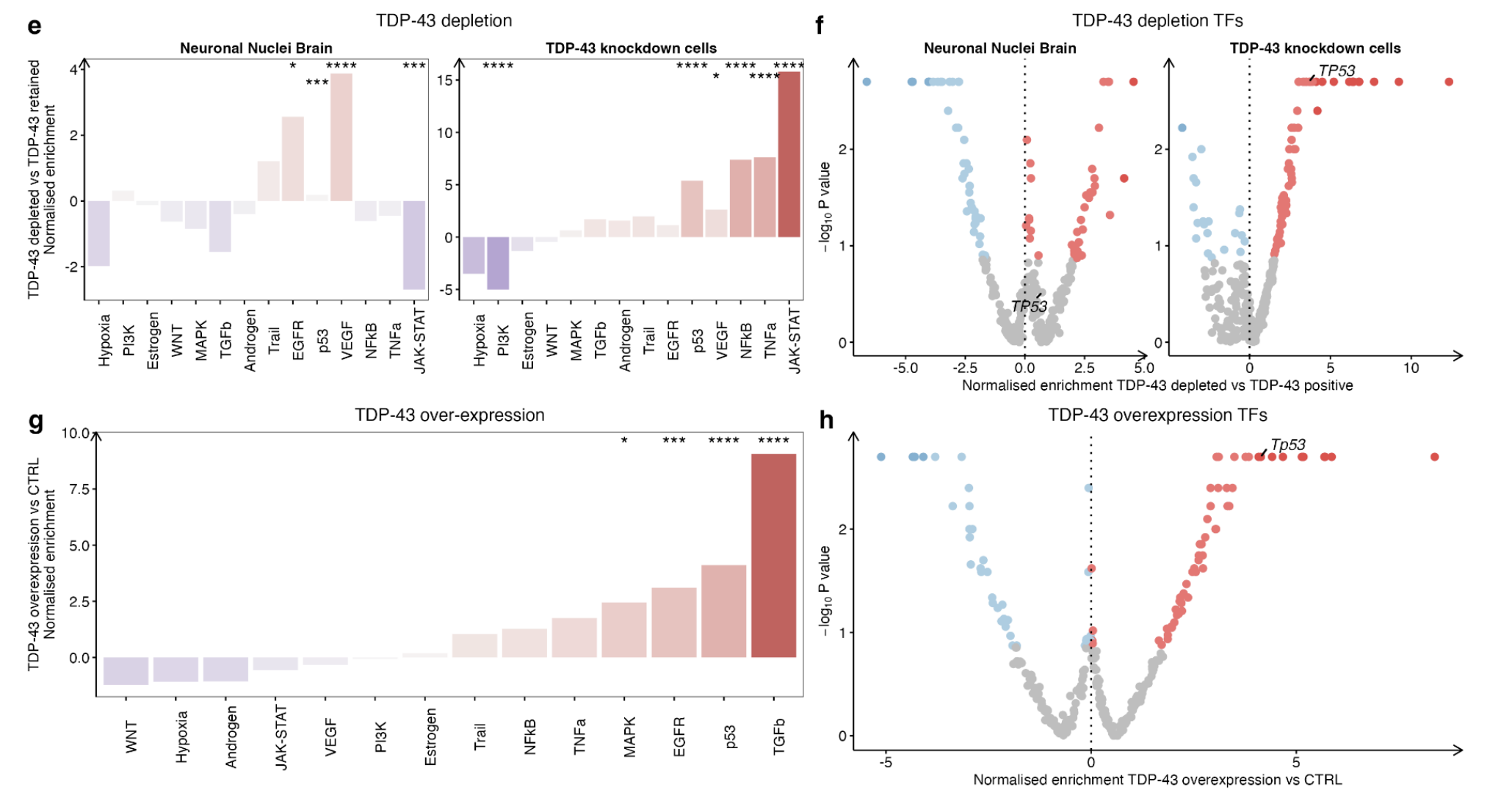
ALS iPSMNs and post-mortem tissue exhibit extensive alterations in splicing
An increasingly proposed mechanism of disease in ALS is dysregulated alternative splicing, which may also contribute to genomic instability.
MAJIQ (Ref 4)
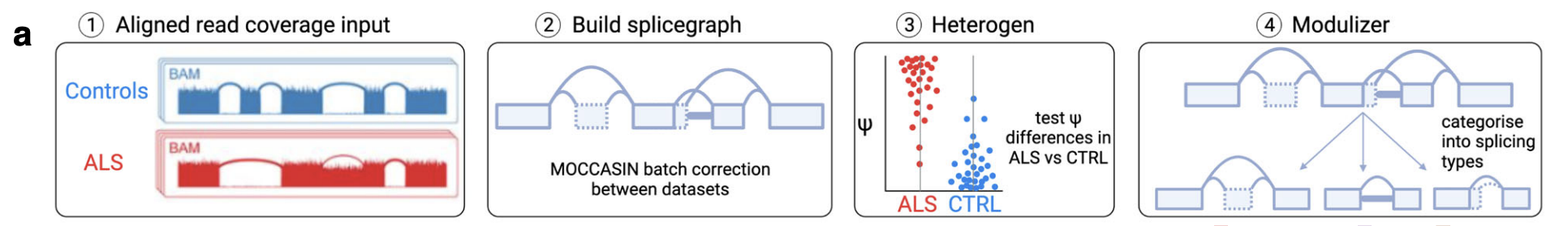
iPSMNs
poly(A) selected libraries(10 datasets composed of 48 ALS and 43 control iPSMNs)
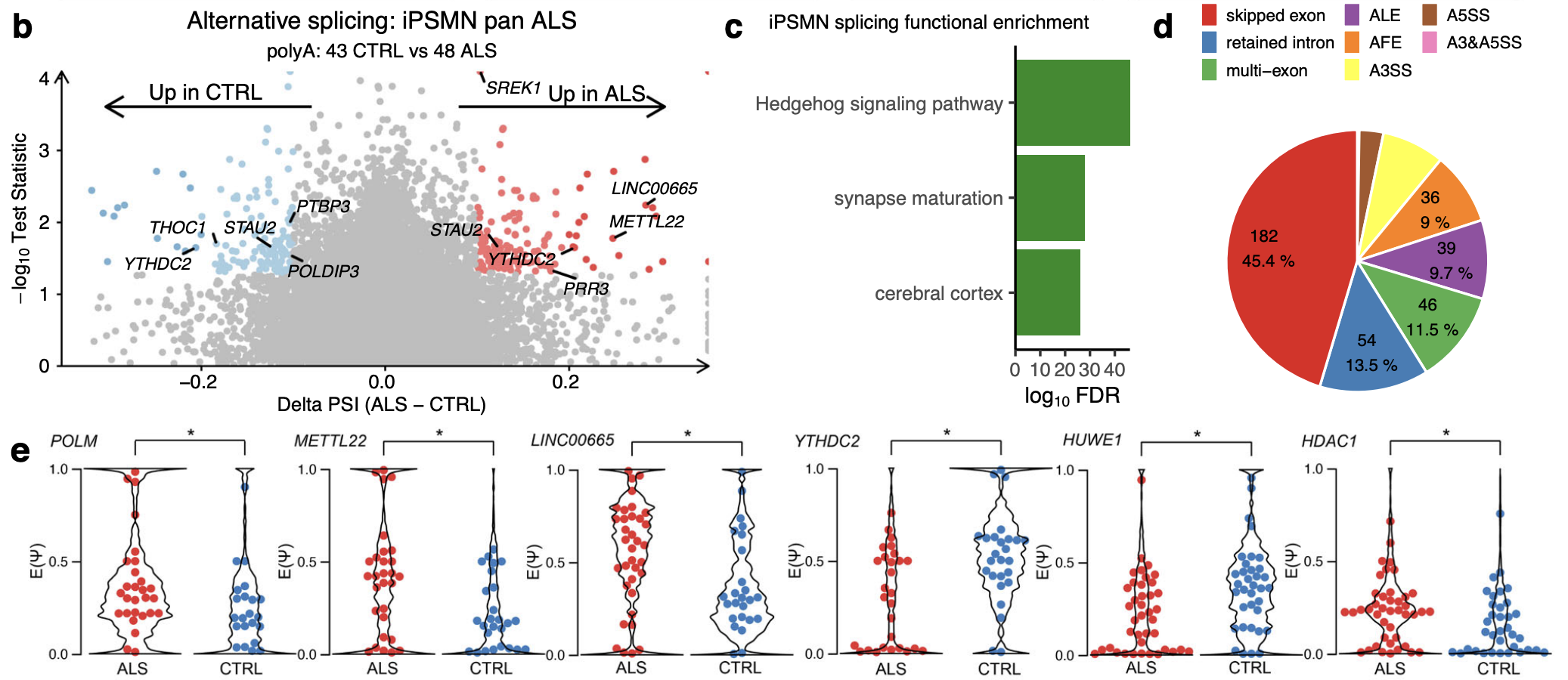
ALS versus control iPSMNs
Comparing ALS vs control iPSMNs identified 264 local splice variation events in 161 unique genes that were significantly different between ALS and control.(TNOM p < 0.05, Δ PSI > 0.1; Fig. 5b)
Tips: Alternative splicing events are quantified in terms of percent spliced in (PSI, denoted by Ψ), which is the relative ratio of isoforms including a specific splicing junction or retained intron. There are two main modes of executing the quantifier: Quantifying the relative inclusion levels of LSVs in a given experimental condition (also known as “percent selected index”, PSI, or Ψ), and quantifying changes of LSVs inclusion levels between two experimental conditions (aka delta PSI or ΔΨ).
Amongst the genes exhibiting differential splicing in ALS were a significant number of genes involved with p53 and DNA repair (including POLM, METTL22, HUWE1, HDAC1, MTA1, PMS1, ZSWIM7; Fisher exact test p = 2.3 × 10−8).
Likewise, there was a greater number of RBPs amongst differentially spliced genes than expected by chance (e.g. YTHDC2, THOC1, PRR3, STAU2, PTBP3, SREK1, POLDIP3; p = 6.8 × 10−18; Fig. 5e).
Functional over-representation analysis of the 161 genes containing differential splicing showed enrichment in protein binding (FDR = 0.02), synaptic (FDR = 0.03) and neuronal functions (FDR = 0.04, Fig. 5c), which are central to ALS motor neuron pathophysiology.
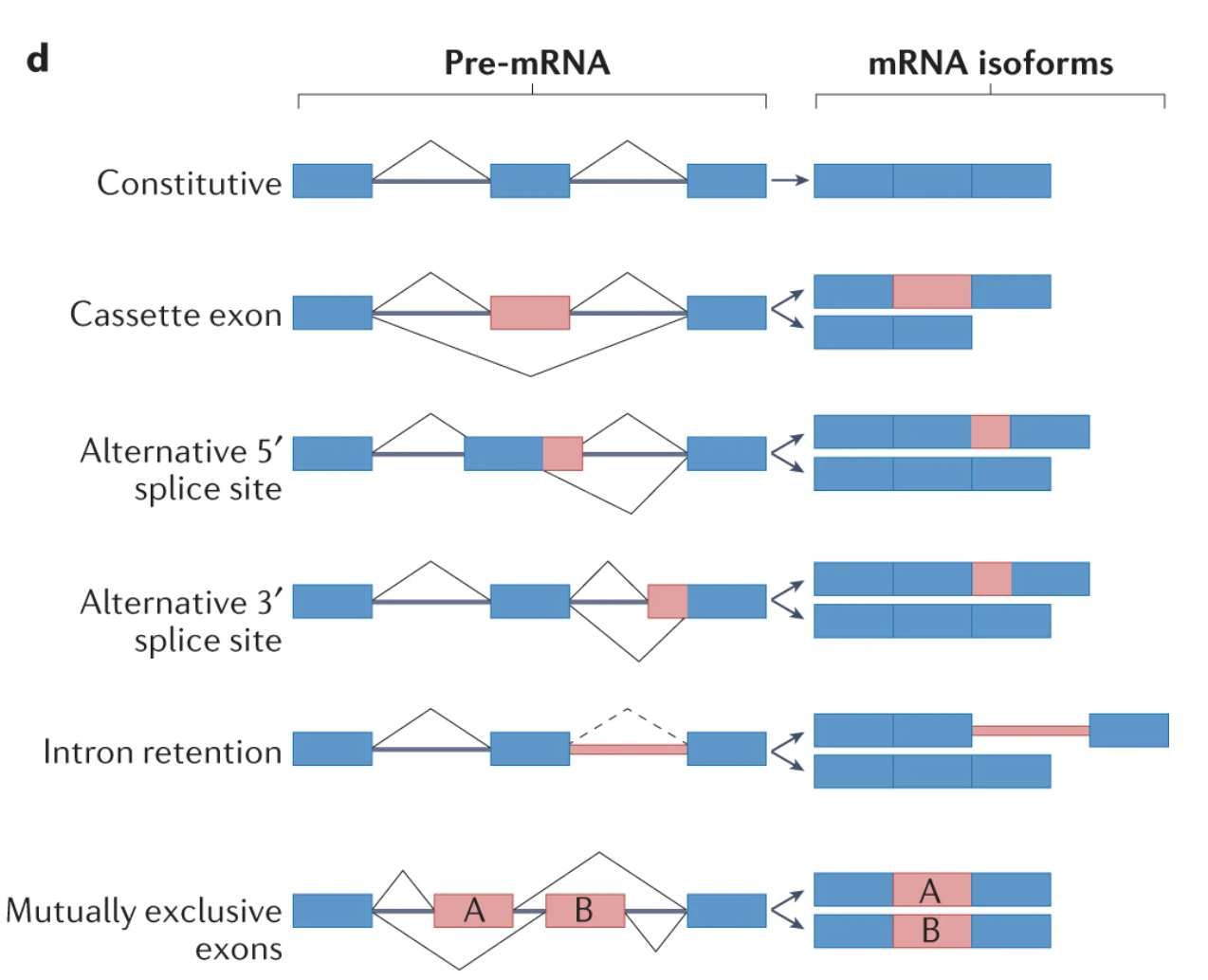
The local splice variations types (Fig. 5d):
Furthermore, splice events are not restricted to annotated reference transcriptome splice sites and of the 264 differential splicing events, 28 (10.6%) involved de novo splice junctions. Of these, 7 were found to be cryptic exons (RELCH, HOXC4, RBM26, SLC35B3, TENM3, TPTEP2-CSNK1E, ZSCAN29) although none of these overlapped with TDP-43 depletion. 50 out of 264 (18.9%) differential splice events harboured IR within the local splice variation.
Breaking down each local splice variation into its component splice types and categorising these into basic splicing modules revealed that exon skipping was the most common splicing type (182, 45.4%) followed by IR (54, 13.5%; Fig. 5d).
Each ALS genetic subgroup
We next investigated alternative splicing in each ALS genetic subgroup separately. Whilst there were no sporadic ALS iPSMNs that had undergone poly(A) selection, there were 23 C9orf72, 9 FUS, 9 TARDBP and 4 SOD1 mutant poly(A) samples.
Compared to controls, TARDBP mutants showed the greatest number of differential splicing events (1435), followed by FUS (1099), C9orf72 (429) and SOD1 (256).
Functional over-representation analysis in each mutant group revealed that genes exhibiting differential splicing were involved with protein binding, neuronal structures, and RNA processing (Supplementary Fig. 17e–h).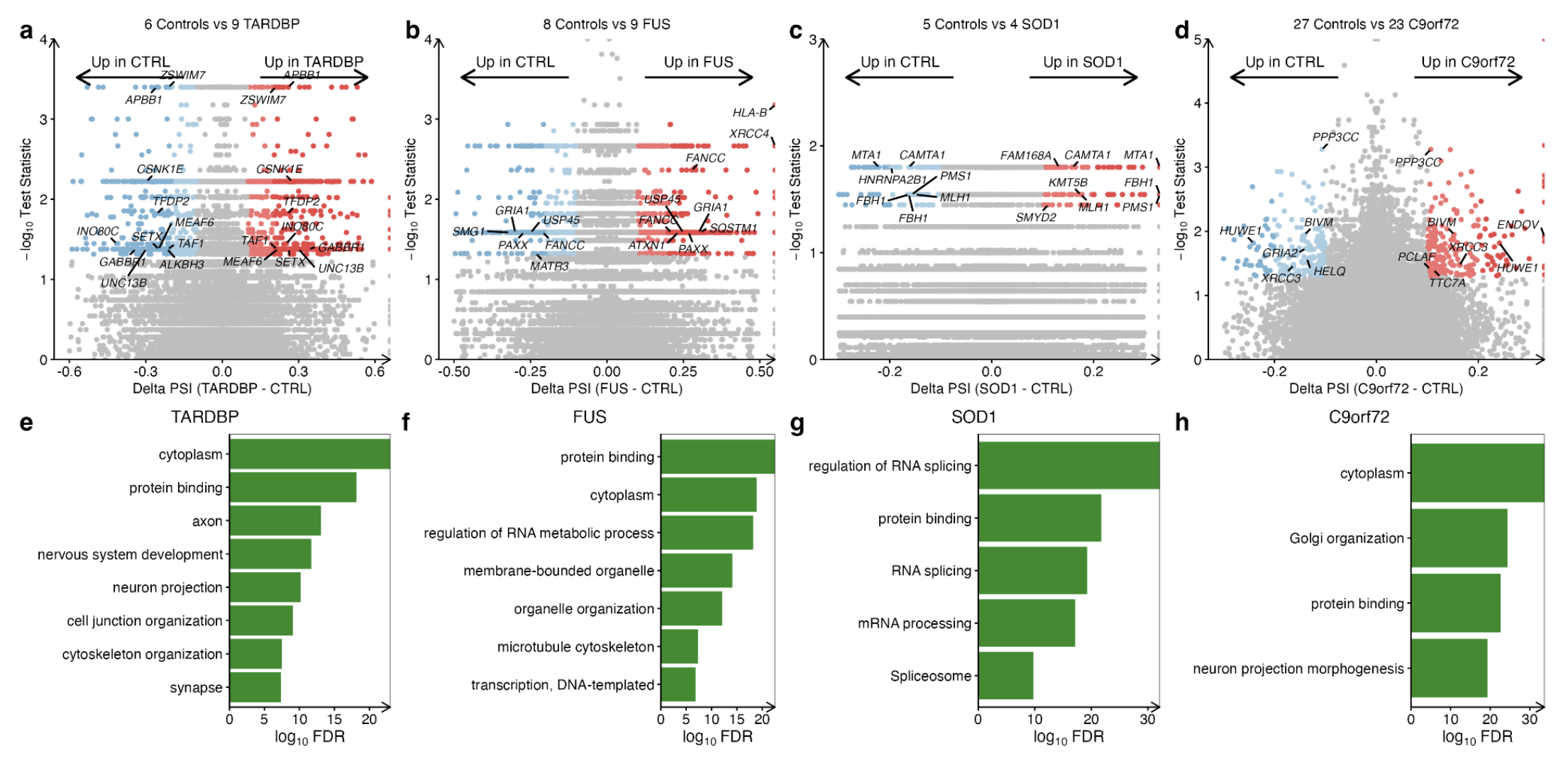
The local splice variations types: Exon skipping was the most common splicing type in TARDBP, FUS and C9orf72 subgroups, however, in SOD1 mutants, intron retention was the most frequent (Supplementary Fig. 17i).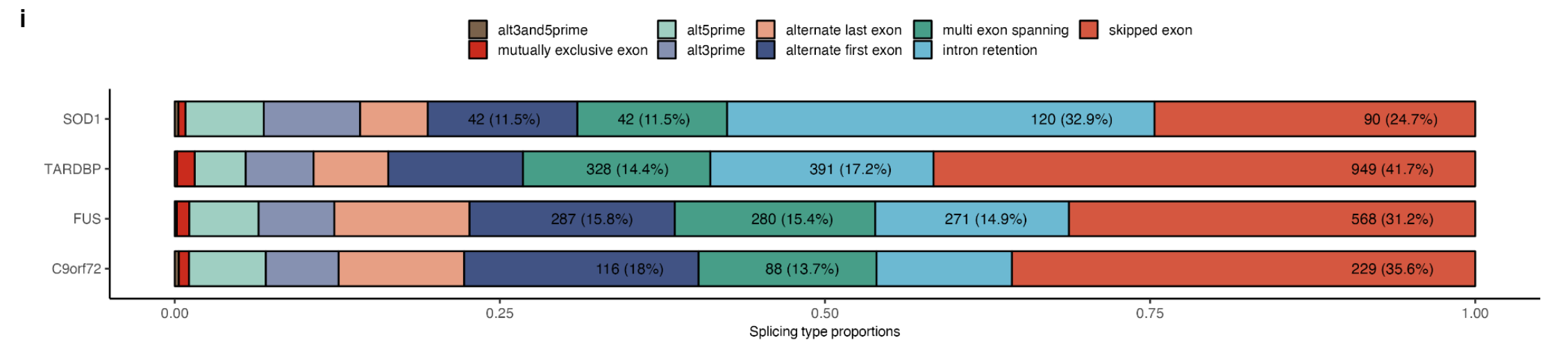
Pearson Correlation: Correlation of alternative splicing changes between genetic subgroups revealed weak associations with the strongest correlation between FUS and C9orf72 mutations (R = + 0.09) and the weakest correlation between SOD1 and TARDBP (R = −0.1; Supplementary Fig. 17j).
Intersecting significant splicing changes between genetic backgrounds revealed that none were common to each genetic group, but that 33 were shared amongst two mutant groups. Amongst these were the p53 signalling and RBP gene CNOT3 as well as other RBPs including SRSF10, DNAJC17, SRRM1, SIDT2, and SREK1.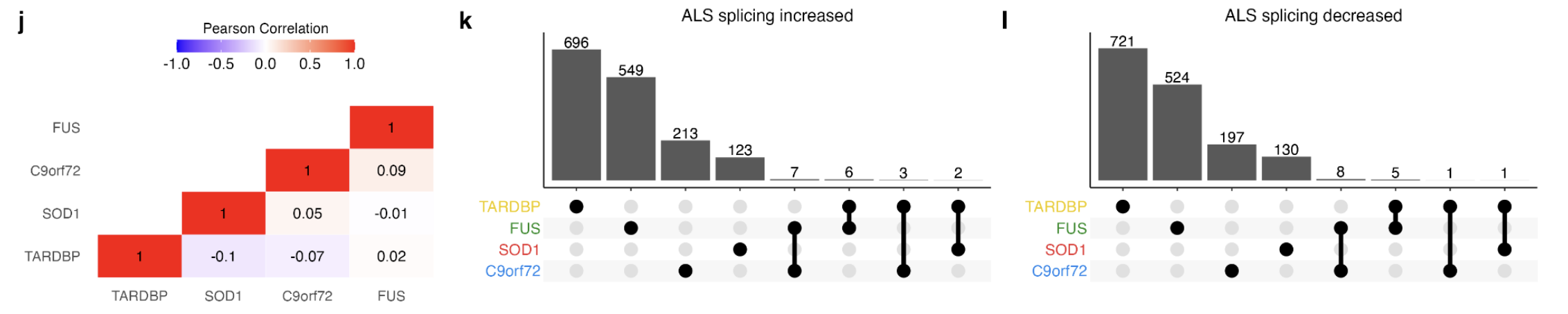
FACS-sorted neuronal nuclei (TDP-43 depletion)
By examining splicing changes in neuronal nuclei depleted of TDP-43, researchers found 12 overlapping differentially spliced genes (encompassing 17 splicing events) with ALS iPSMNs (including POLDIP3, PPP6R3, CAMK2B, CEP290).
Tips: POLDIP3 encodes an RRM (RNA recognition motif)-containing protein that participates in the regulation of translation by recruiting ribosomal protein S6 kinase beta-1 to mRNAs. Alternative splicing results in multiple transcript variants.
TDP-43 knockdown
Similarly, comparing splicing changes upon TDP-43 knockdown with ALS iPSMNs revealed 4 overlapping genes containing 6 splicing events (POLDIP3, CAMK2B, HERC2P3, CEP290;).
Interestingly, the multi-exon skipping splicing event in POLDIP3 was precisely the same event that occurs in both TDP-43 neuronal nuclei depletion and TDP-43 knockdown.
This indicates that TDP-43 nuclear loss of function may contribute to splicing changes in ALS iPSMNs.
Postmotrem cohort (NYGC cohort)
Because of RNA degradation in post-mortem tissue, the NYGC samples were generated using ribosomal depletion instead of poly(A) library selection.
Comparing splicing in post-mortem ALS versus control samples revealed 842 significant local splice events in 445 unique genes (Δ PSI > 0.1, tnom p < 0.05; Fig. 5f).
Amongst the differential splicing events in ALS post-mortem were 4 established ALS genes (CAMTA1, NEK1, ATXN1, and GRIN1), 19 genes with altered splicing in neuronal nuclei depleted of TDP-43 (including KALRN, PRUNE2, DNM1) and 11 genes with altered splicing in TDP-43 knockdown (e.g. KALRN, DNM1, NEK1; Fig. 5i, Supplementary Data 11).
A significant number of genes that encode DNA damage repair factors (e.g. APTX, CENPX, RIF1, CNOT3; Fisher p = 6.7 × 10−16) and RBPs (e.g. EIF4E3, HNRNPUL1, ATXN1, SRSF5; Fisher p = 1.3 × 10−26).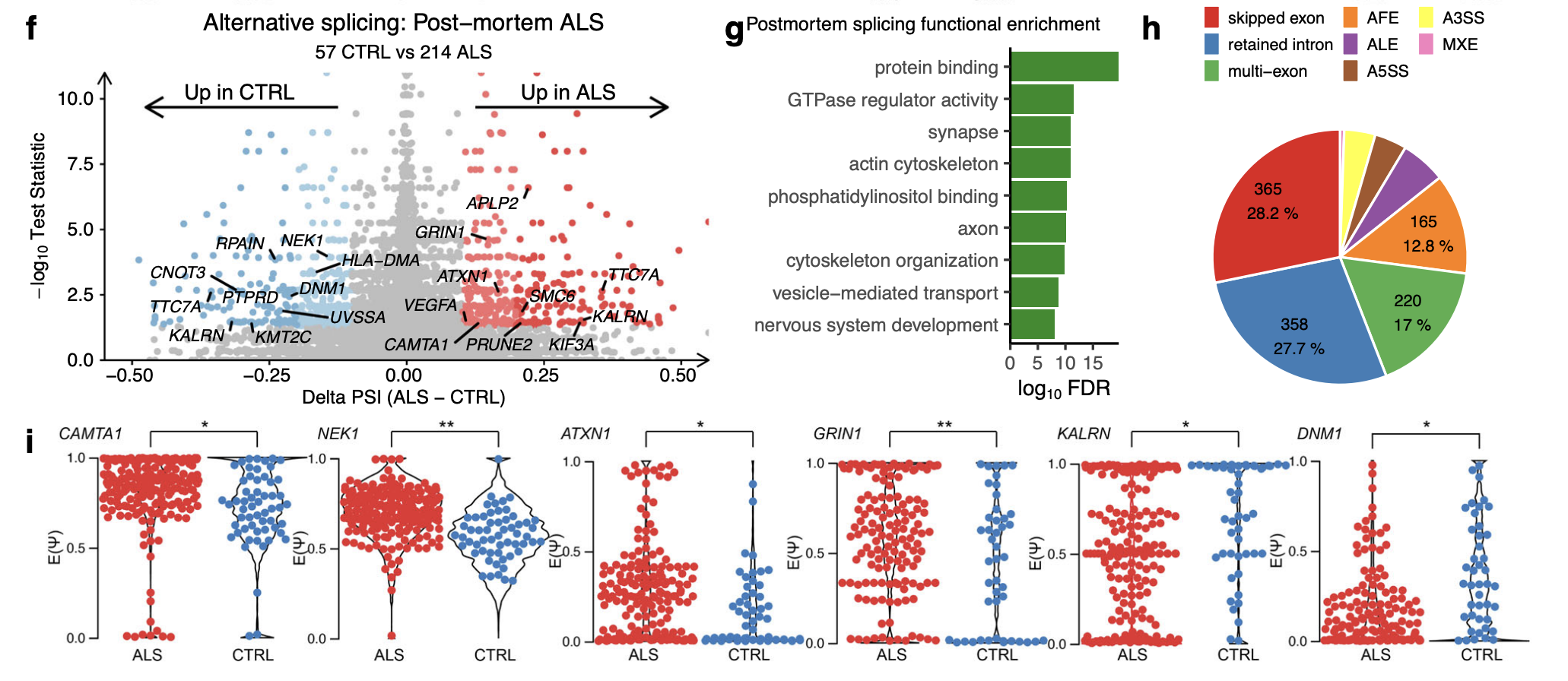
Functional enrichment analysis (Fig. 5g):
protein binding (FDR = 2.5 × 10−10)
neuron compartments (FDR = 1.6 × 10−6, Fig. 5g).
The local splice variations types:
Of the 842 differential splicing events, 178 (21.1%) involved de novo splice junctions and of these, 21 were cryptic exons of which EP400, PLEKHA1, BMP2K, and KMT2C overlapped with TDP-43 depletion.
IR was the second most common splicing type, accounting for 27.7% of all post-mortem splicing events, behind skipped exons (28.2%; Fig. 5h).
Somatic mutation burden in ALS iPSMNs and post-mortem tissue
Genome instability triggers the DNA damage response and p53 signalling.
iPSMNs
GATK variant discovery pipeline ( detects singlenucleotide variants (SNVs), insertions and deletions (indels))
Only Answer ALS dataset
All filtered variant types were significantly greater numbers of somatic mutations per iPSMN in ALS compared to control (Wald test p < 2 × 10−16; Fig. 6a).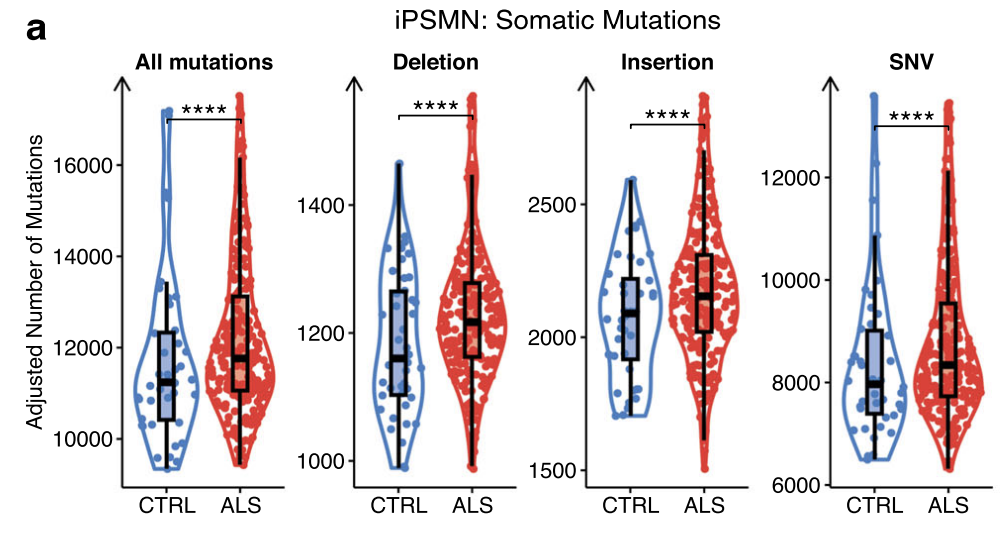
Assessing the relative contributions of each base substitution type revealed largely similar SNV spectrum profiles in ALS and CTRL iPSMNs, predominantly composed of T > C followed by C > T substitutions.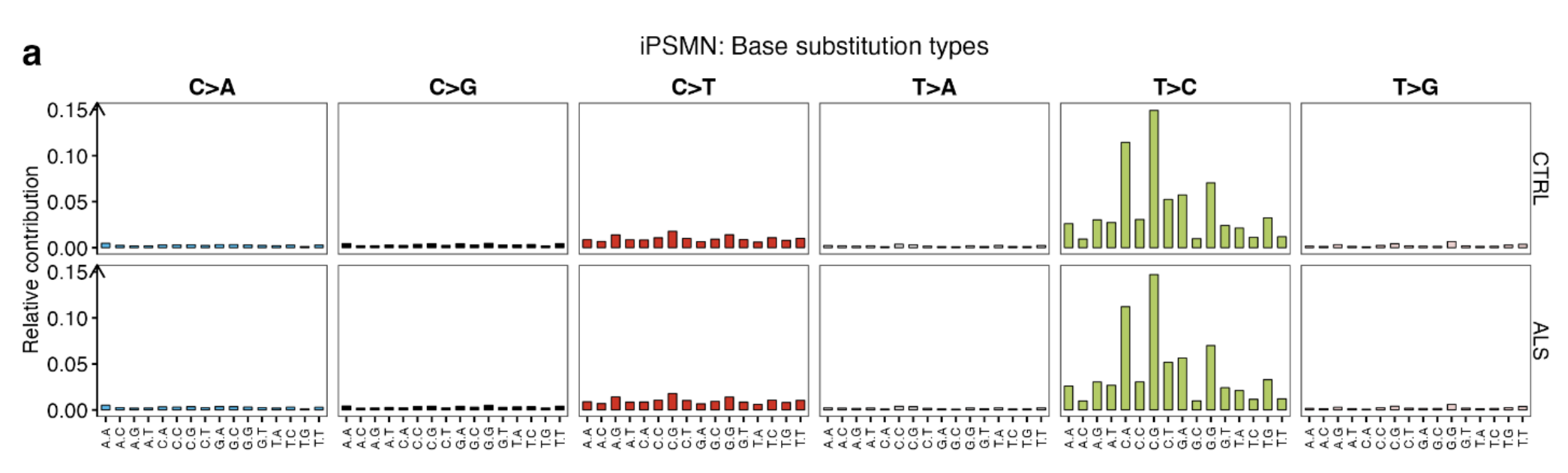
Examining each genetic background separately revealed that sporadic iPSMNs showed significant increases in SNVs (p < 2 × 10−16), insertions (p = 5.7 × 10−15), and deletions (p < 2 × 10−16), C9orf72 mutants showed significant increases in SNVs (p < 2 × 10−16) and insertions (p = 4.3 × 10−10), whilst SOD1 mutants showed significant increases in insertions only (p = 7.3 × 10−10; Fig. 6b).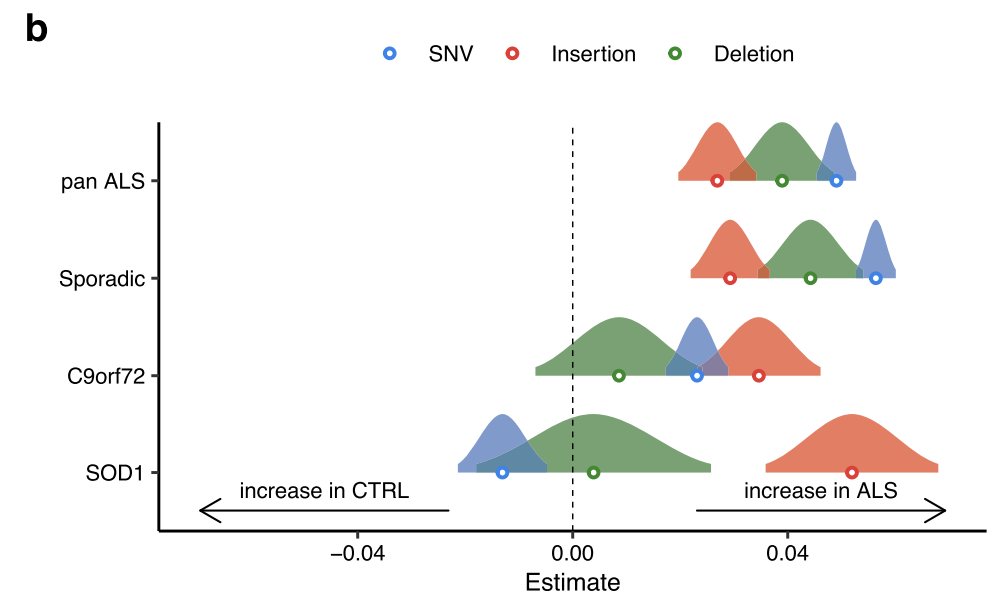
Forest plot showing the generalised linear model point estimate and 95% confidence interval of changes in mutation types (SNV, blue; insertion, red; deletion, green) in ALS genetic subgroups versus controls. The vertical dashed line indicates no difference, to the right of the dashed line indicates an increase in ALS.
Postmortem
As iPSMNs are derived from peripheral cell types and the reprogramming process itself can induce somatic mutations, genome instability in iPSMNs may not be representative of cell types in the central nervous system.
All filtered variant types were significantly greater numbers of somatic mutations
Same relative contributions of each base substitution type: T>C substitutions followed by C > T substitutions
Each genetic background: SOD1 mutants showed significant increases in insertions and Deletion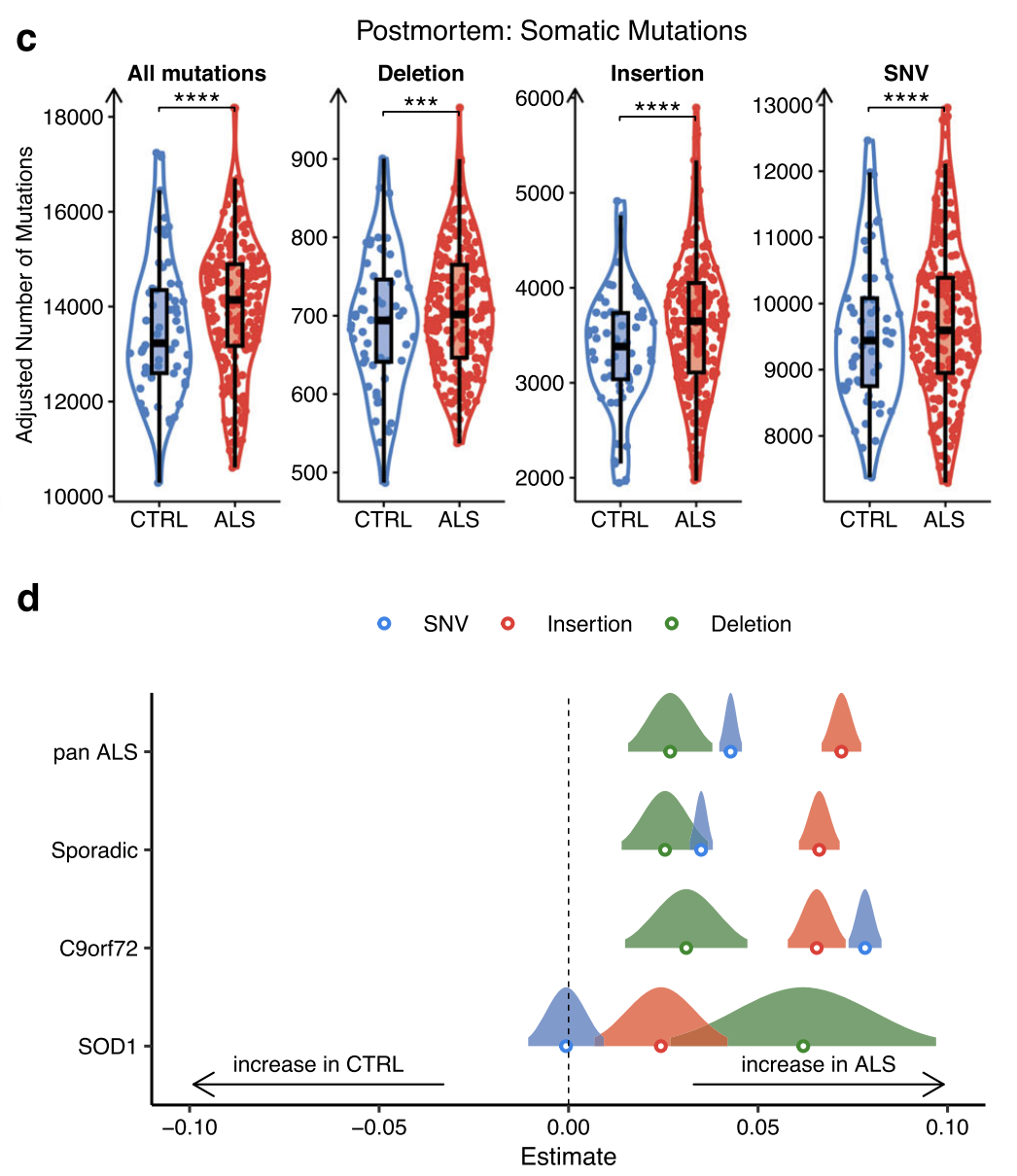
This greater burden of somatic mutations in ALS may contribute to the heightened DNA damage response and implicates defective DNA damage repair in ALS.
Landscape of gene fusions across ALS
Gene fusions are another important class of genome alteration that can arise from the repair of damaged DNA. Fusions involve two genes becoming juxtaposed due to genomic structural rearrangements, including inversions and translocations.
STAR fusion pipeline
iPSMN
11 paired-end RNA-seq iPSMN datasets
292 unique gene fusions in ALS iPSMNs and 152 unique gene fusions in control iPSMNs, with 91 shared in both conditions. Among the 201 gene fusions identified in ALS iPSMNs but not in controls, were fusions affecting genes implicated in ALS including VAPB–APCDD1LDT, ATXN1–ZFYVE27, TUBA1A–NEFM and OSTF1–APP.
Furthermore, of the 292 gene fusions identified in ALS iPSMNs, 14 affected genes also exhibited altered splicing, supporting the possibility of trans-splicing, that post-transcriptionally joins exons from separate pre-mRNAs.
Burden analysis: By comparing the proportion of each unique gene fusion in ALS with CTRL iPSMNs, we identified 9 gene fusions with a significantly greater burden in ALS iPSMNs (Supplementary Data 14). These mostly involved long noncoding RNAs (lncRNAs), for example, the gene fusion with the greatest burden in ALS was a neighbour fusion between the lncRNA LINC01572 and PMFBP1 (OR 3.3, 95% CI 1.6-Inf, Fisher’s exact test p 0.001).
Significantly greater numbers of gene fusions in ALS compared to control samples
Examining each genetic background:
significantly greater numbers of gene fusions in C9orf72 and SOD1 mutant groups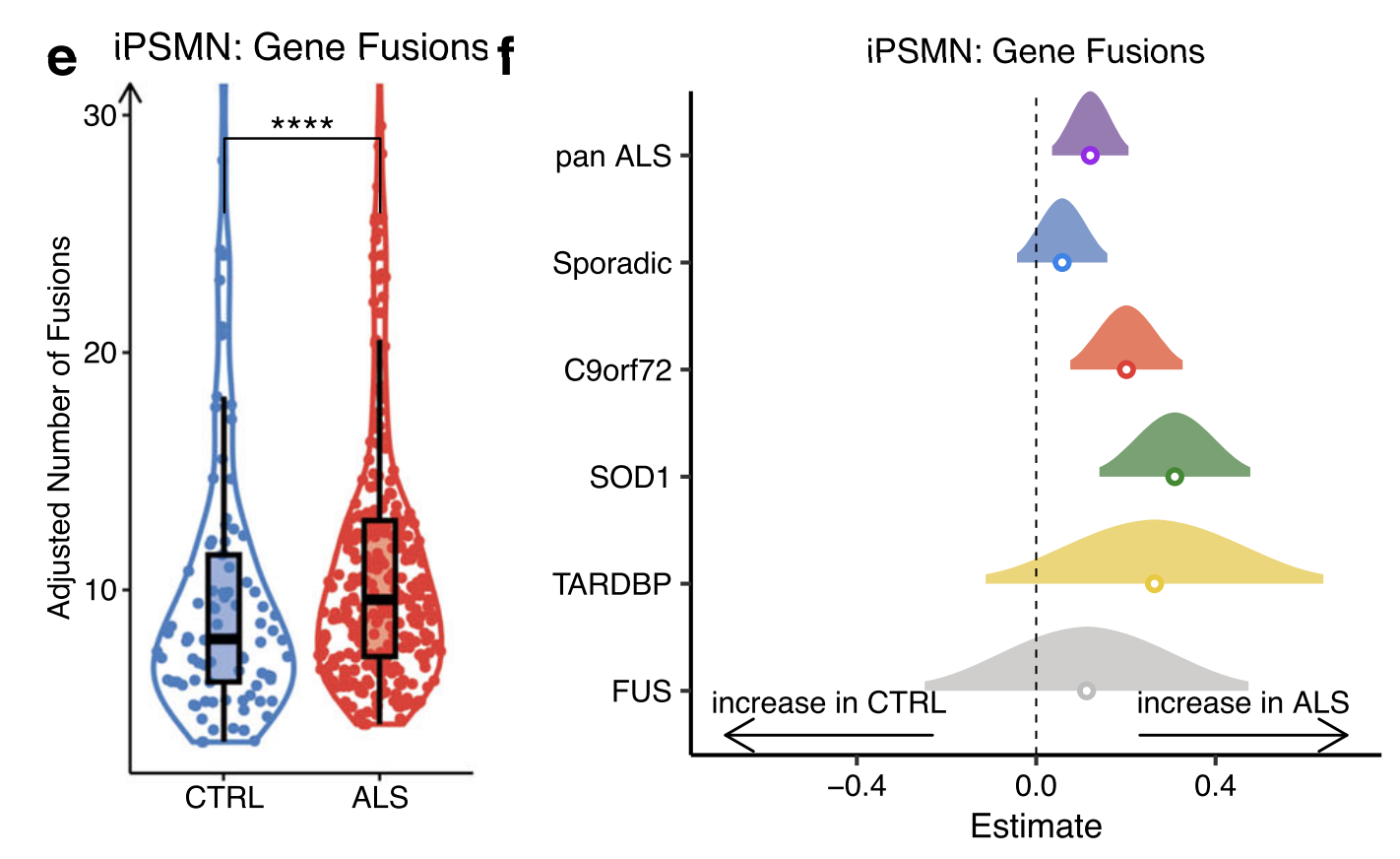
Fusion Types: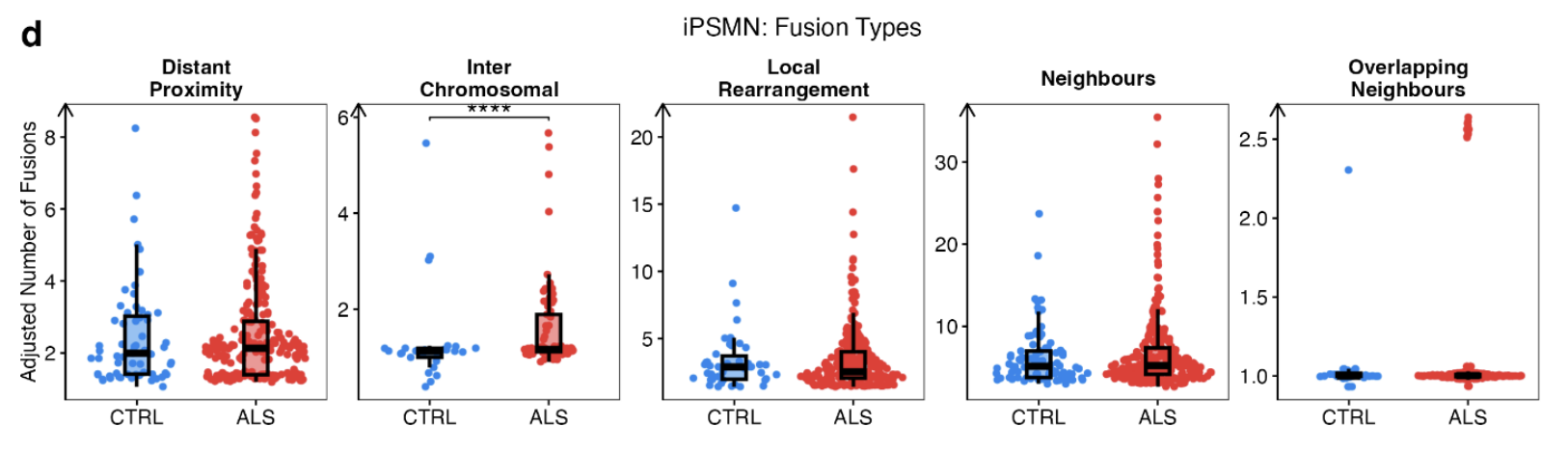
Postmortem
There were a total of 177 unique gene fusions in ALS and 96 in control post-mortem, with 71 shared.
Significantly greater numbers of gene fusions in ALS compared to control samples
Burden analysis: identified 13 gene fusions with a significantly greater burden in ALS post-mortem tissue. As with iPSMNs, these mostly involved lncRNAs and the fusion with the greatest burden in ALS post-mortem was a gene neighbour fusion between the lncRNA AL353138.1 and PTCHD4.
Each ALS genetic group: revealed significantly greater numbers of gene fusions in sporadic ALS (p = 8.2 × 10−6), C9orf72 (p = 0.03) and SOD1 mutant subgroups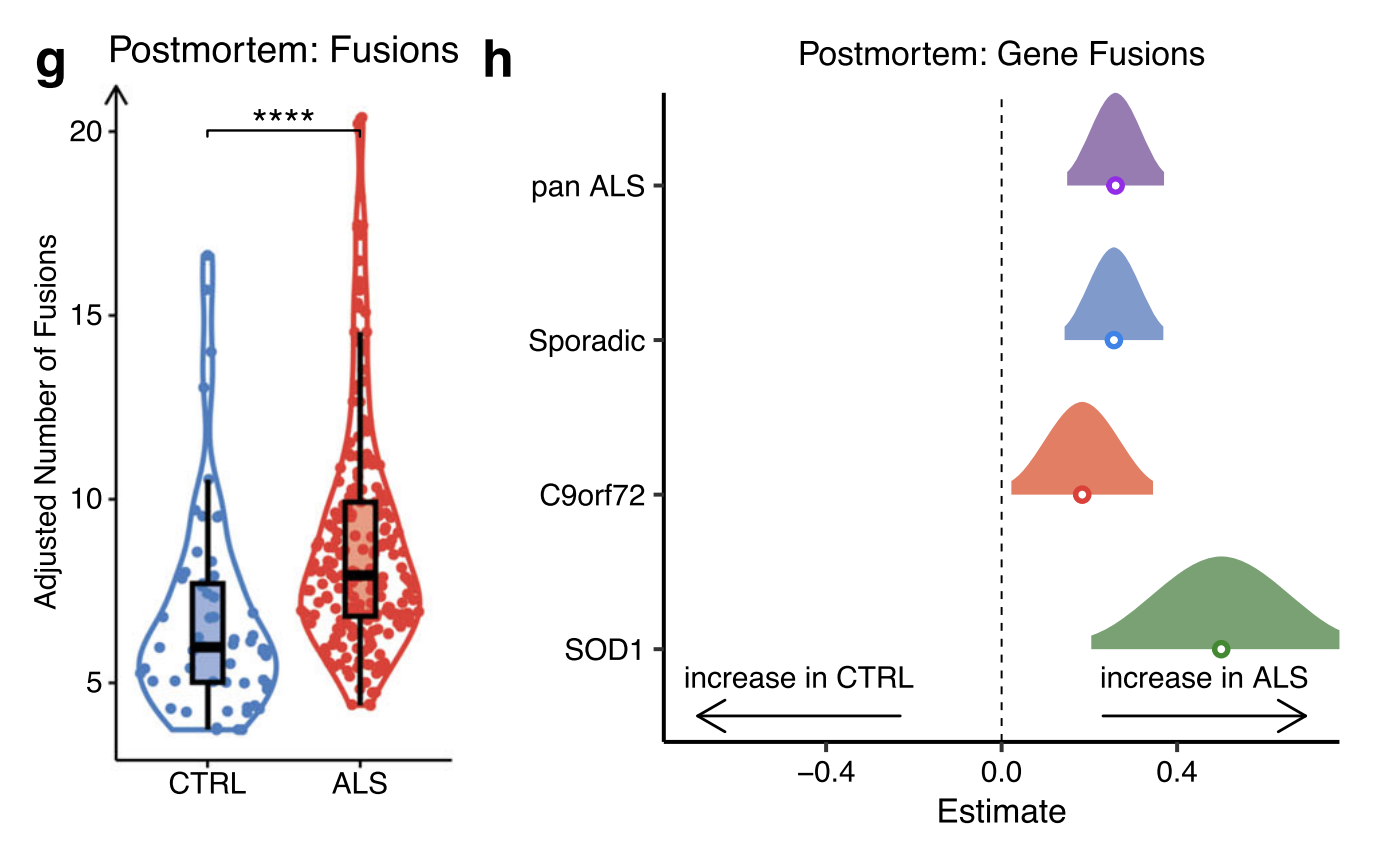
Taken together, these findings reveal enrichment of SNVs, indels and gene fusions in ALS iPSMNs and post-mortem tissue, it is likely to be a genomic signature arising from elevated DNA damage and/or impaired DNA repair.
Discussion
p53 activate in ALS, particularly with C9orf72 repeat expansions. p53 activation was accompanied by increased somatic mutations and gene fusions across diverse ALS subgroups, in both iPSMNs and post-mortem tissue. This result consistent with reports that C9orf72 repeat expansion induces DNA damage, likely mediated by dipeptide repeat proteins and the formation of R loops and G quadruplexes.
Highlight: p53 was strongly and significantly activated in TARDBP and sporadic subgroups. This finding in sporadic cases is particularly important since they represent ~80% of ALS cases, but have the least prior evidence for p53 activation.
In lack TDP-43 pathology (FUS and SOD1 mutants) cases, the p53 activation was weaker than that in TDP-43 ALS cases, raising the possibility that TDP-43 contributes to the DNA damage response. Together, these results suggest that TDP-43 pathology exacerbates the DNA damage response, and subsequent p53 activation may promote motor neuron death in ALS.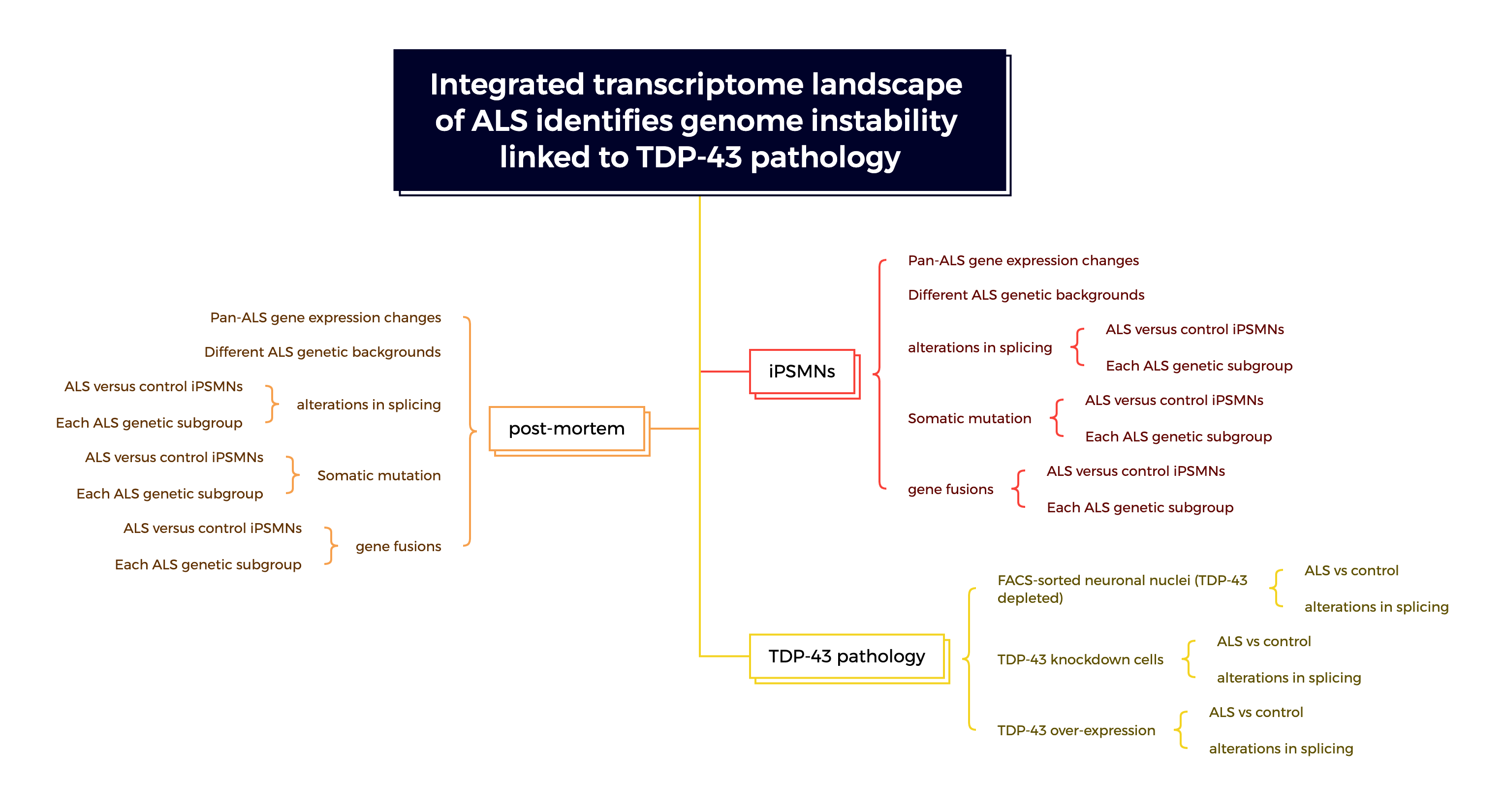
Limitation
PROGENy and DoRothEA weights are based on perturbation experiments that are not specific to motor neurons. Their signalling pathways may activate diverse downstream gene expression programmes depending on the cell type and perturbing agent utilised.
Environmental risk factors play an important role in ALS aetiology, and while the iPSC model is an elegant approach to model sporadic ALS, a notable limitation is that it does not reproduce patients’ environmental exposures or aging signatures
Reference
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. The Lancet. 2022 Oct;400(10360):1363–80.
- Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun. 2018 Jan 2;9(1):20.
- Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature. 2022 Mar 3;603(7899):124–30.
- Vaquero-Garcia J, Aicher JK, Jewell S, Gazzara MR, Radens CM, Jha A, et al. RNA splicing analysis using heterogeneous and large RNA-seq datasets. Nat Commun. 2023 Mar 3;14(1):1230.
- Marasco LE, Kornblihtt AR. The physiology of alternative splicing. Nat Rev Mol Cell Biol. 2023 Apr;24(4):242–54.