Proteomics
Proteomics info collection
Bottom-up proteomics
The system-wide characterization of the proteome using mass spectrometry (MS), and more specifically on bottom-up proteomics where proteins are digested into smaller pieces called peptides, which are analysed by MS.
MS-based proteomics can analyse the protein content of any material. Apart from mainstream sources such as cell lines, this encompasses clinically important, archived formalin-fixed paraffin-embedded (FFPE) biopsy tissues and even fossils that are hundreds of thousands of years old.
Data acquisition
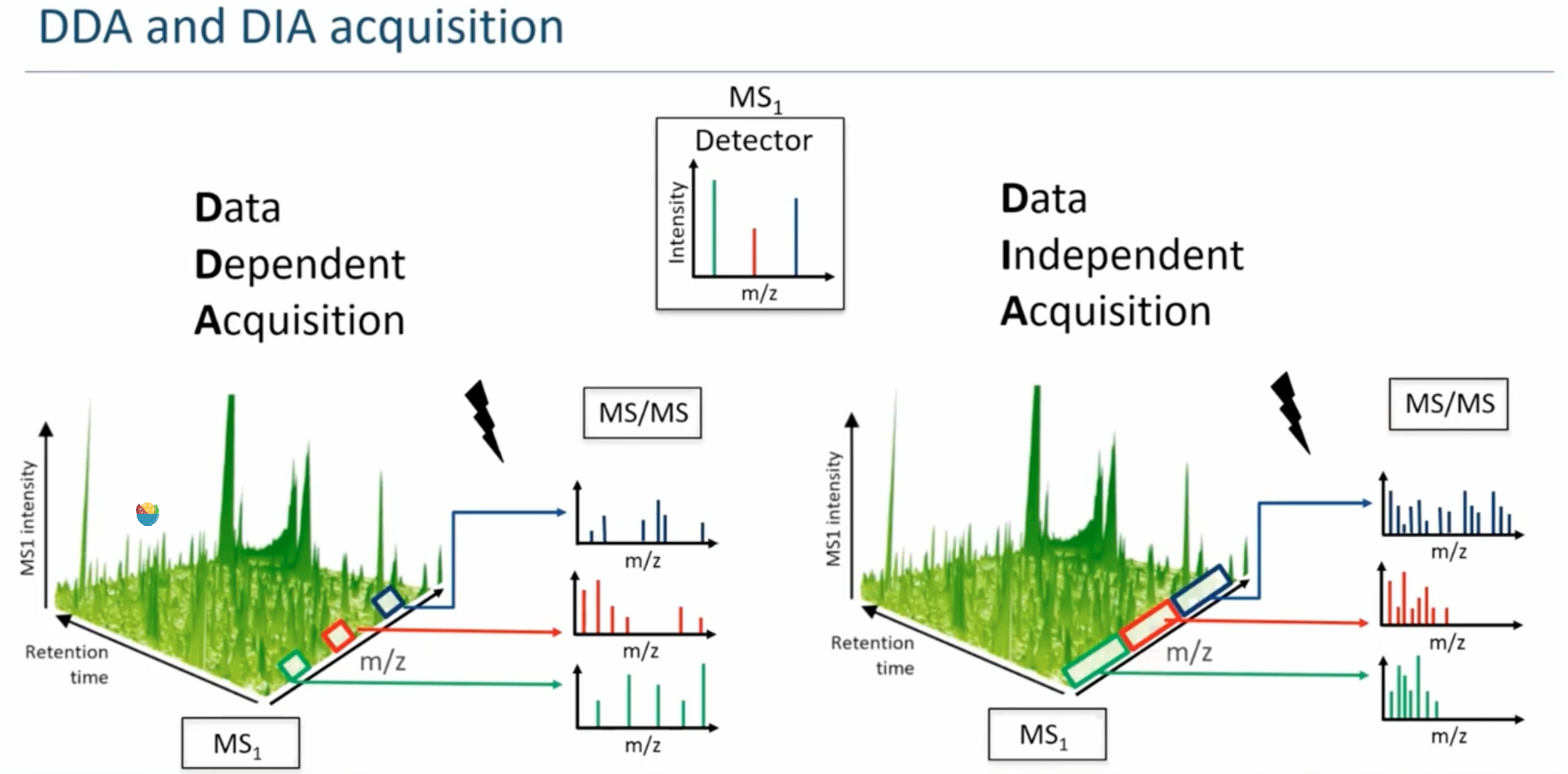
DDA and DIA are the common data acquisition strategies in shotgun proteomics.(how to select picks from MS1 spectrum)
- data-dependent acquisition (DDA), meaning that the mass spectrometer follows a set of user-defined rules (such as m/z, charge, intensity and cross-section) to select as many peptides as possible for acquiring MS/MS spectra. However, this selection is partly stochastic as there are more peptides than analysis time, and it generates missing values.
- In data-independent acquisition (DIA) methods, the quadrupole instead continuously cycles across the entire mass range while selecting relatively large m/z values (20–40 m/z). This leads to very complex MS/MS spectra since they contain the superimposed fragmentation patterns from co-isolated peptide ions. Modern software can deconvolute the spectra to identify the multiple peptides, usually by comparison to a previously acquired ‘peptide library’, but increasingly also without.
Quantification strategies for peptides
- Label-free quantification (LFQ): the MS signals of the peptides (usually at the MS1 level) are extracted from the raw data, normalised and compared between the proteomic conditions of interest. It is experimentally the most straightforward and usually the most economical approach, providing great flexibility in project design. However, this strategy has higher quantification variance, and differences in peptide purity and instrument performance may impact comparisons between individual samples if sufficient care is not taken.
Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ
The MaxLFQ algorithm is integrated into MaxQuant and it is initially developed for DDA (and also can be used for DIA).
One reason for the popularity of MaxLFQ is that it addresses major pitfalls in proteomics quantification by accounting for the fact that different peptides belonging to the same protein can have very different base intensities, for example, due to differing ionization efficiencies. In addition, it is robust against missing values for normalization (differing sample depths) and can reliably estimate protein intensities. - label based: Label-based approaches use stable isotopes to encode different proteome states – the beauty is that the resulting peptides have exactly the same physiochemical behaviour, but have predictable differences in mass. The isotopes can be metabolically introduced, which also allows determination of protein turnover; however, chemical labelling with ‘readout’ at the MS/MS level is now much more common. The latter is referred to as isobaric labelling and involves a clever trick in which the mass of the tag remains the same, but the distribution of isotopes in the tag is revealed after fragmentation. Within a set of 6–16 different tags, quantification variance is typically lower than in LFQ if samples are consistently and reproducibly labelled and combined. A major caveat of the typically used TMT (tandem mass tag) isobaric labelling method is that co-fragmented peptides can suppress quantitative differences (‘ratio compression’). iTRAQ (Isobaric Tags for Relative and Absolute Quantitation) and TMT (Tandem Mass Tags) are two widely used peptide labeling techniques for quantitative proteomics.
tools for DDA LFQ analysis
- LFQ-AnalystThis is a software that supports Label-Free Proteomics Data Preprocessed with MaxQuant.
Normalisation methods
- vsn: Huber W, Von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002 Jul 1;18(suppl_1):S96–104.
MaxQuant summer school
Proteomics
https://xuan-98-l.github.io/2024/08/20/Proteomics/