CytoTRACE文献阅读
CytoTRACE的文献笔记
基础信息
- Title:
Single-cell transcriptional diversity is a hallmark of developmental potential - Author: Gunsagar S. Gulati1
- Year: 2020
- Journal: Science
- IF: 56.9
研究主题
Key theme: 基于scRNA-seq预测细胞分化轨迹的软件。
Summary:
- 无监督(无需先验知识)
- 基于“在分化过程中,转录多样性,即细胞中表达的基因数量,在分化过程中会减少”的理论
- 可识别与分化相关的基因
Research Bacoground/Problem statement:
尽管已有大量用于预测谱系轨迹的计算方法,但它们通常依赖于(i)推断生物过程的起点(以及方向)的先验知识(比如monocle)和(ii)通过细胞中间状态来重建轨迹(比如Velocity)。
此外,利用现有的计算机方法,很难将具有长期再生潜力的静态干细胞与更特化的细胞区分开来。尽管基于基因表达的模型有可能克服这些限制[例如转录熵(文献11-13)、多能性相关基因集(文献14)和机器学习策略(文献15)],但它们在不同的发育系统和不同的测序平台中的实用性仍不清楚。
基于RNA-based features,包括18,711个带注释的基因集(包含MSigDB,ENCODE和ChEA转录因子,mRNAsi肿瘤细胞Biomarker和三个计算机手段预测干性的StemID,SCENT,Slice),作者确定了不受组织类型、物种和测序平台准确预测细胞分化状态的因素。基于“在分化过程中,转录多样性,即细胞中表达的基因数量,在分化过程中会减少”的理论,作者开发了无监督方法,用于预测单细胞转录组的相对分化状态。整体来说,使用CytoTRACE可以无需先验知识的定义分化起始点,并对一些干细胞相关基因进行排序,识别与干性和分化相关的基因。
Methods:
- 在单细胞数据中筛选细胞分化的关键基因:
- 训练集:使用由 9 个具有实验确认的分化轨迹作为金标准的 scRNA-seq 数据集作为训练集。选择这些数据集是为了优先考虑早期研究中常用的基准数据集,并确保从哺乳动物受精卵到终末分化细胞的发育状态的广泛采样。训练集包含涵盖 2个物种(人和小鼠),49 个表型的 3174 个单细胞、六个组织和三个 scRNA-seq 平台(CEL-seq,Fluidigm C1, Tang在2009年首次报道的scRNA-seq技术)。
- 性能评估:使用 Spearman 相关性将每个基于 RNA 的基因(按表型平均)与已知的分化状态进行比较(图 1A)。然后,对九个训练数据集的结果进行平均,得出每个基因的最终分数和排名。图2B表明该方法揭示了许多已知的,和意想不到的分化状态相关性。
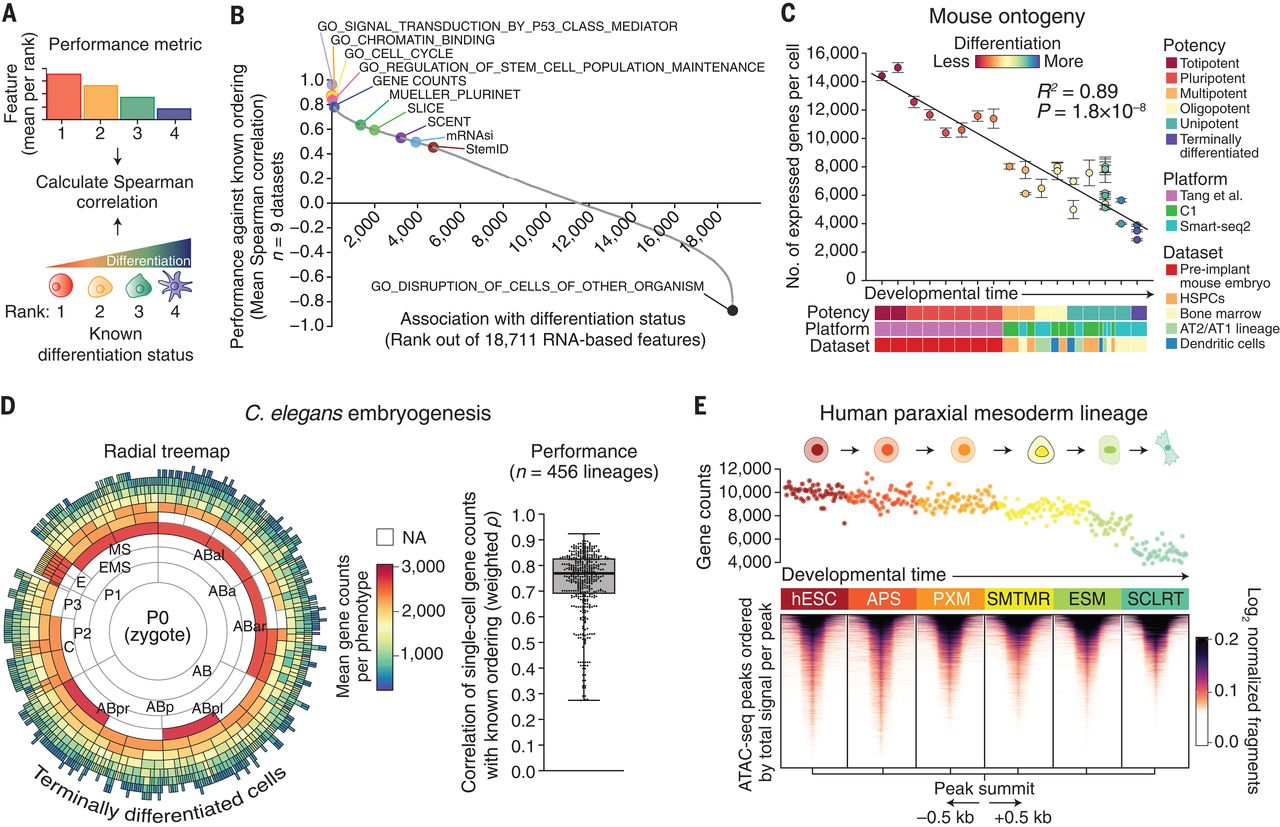
- 结果:发现一个特征显示出显著的性能(104 of 18,711):每个细胞可检测到的表达基因的数量(gene count)(可于上图中2B的蓝点中看到)。由于与成熟的干细胞特征相比,包括细胞周期和多能性基因 (14,15),而且还显示出独特生物学的证据和更广泛的适用性(在cycling cells, noncycling cells, or published data from the earliest stages of human embryogenesis prior to the up-regulation of pluripotency factors中,基因计数都随着分化而减少)。基因计数可能超越孤立的实验系统,在不同细胞类型中概括发育潜力的全部范围。作者在秀丽隐杆线虫(图1D)和斑马鱼(表S4)中做了验证,表明gene count是细胞个体发育的一般特征。
- 补充验证:由于细胞的转录输出与其全基因组染色质谱相关,接下来测试了单细胞基因计数是否最终可以替代全局染色质可及性(全局染色质可及性已被证明会随着分化而减少)。将来自 scRNA-seq 数据的单细胞基因计数与从最近一项人类胚胎干细胞 (hESCs) 体外中胚层分化研究中获得的配对bulk ATAC-seq(图1E)进行比较,发现随着 hESC 分化为近轴中胚层和侧中胚层谱系,观察到全基因组染色质可及性逐渐降低。
- CytoTRACE 的开发:
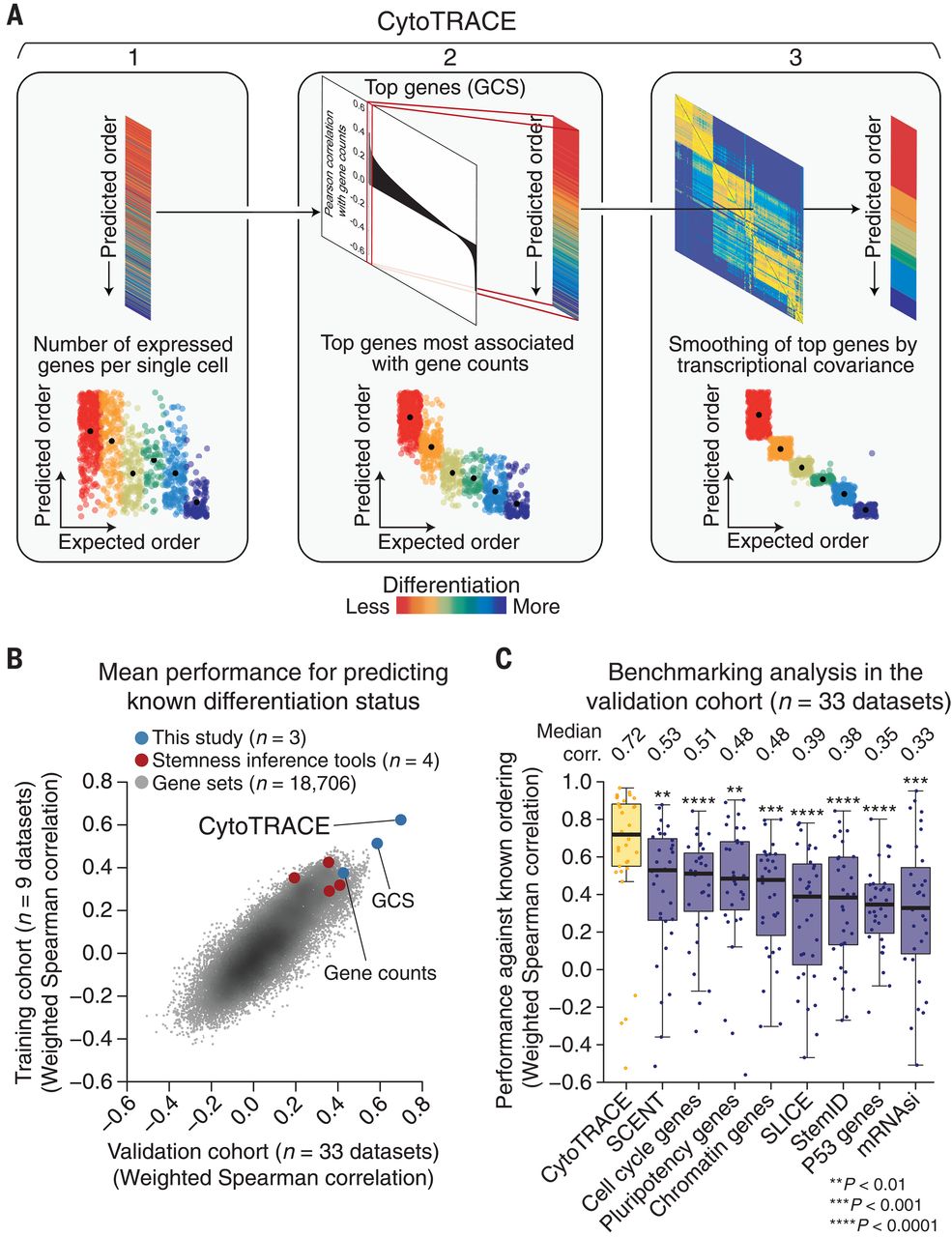
- 已知:每个细胞的表达基因数量通常与RNA丰度呈现正相关,但在部分数据集中,例如 hESC 体外分化为原肠胚层的数据集,每个细胞表达的基因数量表现出相当大的表型内变异(见图2A左)。
- 假设:由于 scRNA-seq 旨在捕获单细胞基因表达,而不是gene count,因此我们推断表达模式与基因计数相关的基因可能会更好地捕获分化状态。
- 验证:在预定义基因集和计算工具的测评中,通过计算每个数据集中基因计数最相关的基因的在每个细胞中表达水平的几何平均数,所得的基因计数特征(gene counts signature,GCS)成为在训练集中表现最好的方法。
- 优化运行速度:为了使性能最大化,与基因计数最正相关的top200基因在每个细胞中的集合表达量的平均值被定义为基因技术特征向量(GCS)。
- 优化噪音:虽然GCS改善了与单细胞分化状态的关联,但它还是存在明显的噪音表型内噪音(见图2A中间)。基于假设“转录相似的细胞有相似的分化状态”,作者实施了一个两步程序,根据单细胞之间的转录协方差直接平滑GCS。由此产生的方法,被称之为 CytoTRACE [全称:用于使用基因计数和表达进行细胞 (Cyto) 轨迹重建分析; https://cytotrace.stanford.edu],优于 GCS 和其评估的其他基于 RNA 的特征:
- 预处理:输入原始counts矩阵,进行基因表达数量的检测,过滤NA值和低表达的基因(少于在5%细胞中表达的基因),利用行总和大于0进行筛选。按照批次对单细胞矩阵进行log2标准化,计算细胞间相关性矩阵(similarity matrix),并基于相关性矩阵计算马尔可夫矩阵。
- 步骤一:计算top 200 GCS, 基于non-negative leaast squares regression(NNLS)获取期望的GCS(基于NNLS建立马尔可夫矩阵与GCS的回归关系)
- 步骤二:基于马尔科夫随机过程对期望的GCS进行迭代,优化矫正期望的GCS向量(即每个细胞中基因表达的几何平均数),指导期望的GCS达到平稳,然后最终按照GCS的大小进行排序,对此数值进行标准化(使其范围为0到1)来获得cytotrace的值。计算每一行基因(每个基因在不同细胞中的表达量向量)与cytotrace的皮尔森相关性。
- 跨组织、物种和平台的性能评估
- 测试集:来自 26 项研究的 33 个额外 scRNA-seq 数据集。这些数据集代表了不同的发育和分化过程,由涵盖 266 个表型的 141,267 个单细胞、九个生物系统、五个物种 和 9 个 scRNA-seq 平台(三个droplet-based 和六个plate-based protocols)。
- 结果:在单细胞水平上进行评估时,CytoTRACE 的表现优于验证队列中所有评估的基于 RNA 的特征(图 2B)。在组织类型、物种、分析的细胞数量、时间序列实验与发育状态、或基于板的技术与基于液滴的技术方面,没有观察到显着的性能偏差。
- 与 RNA velocity的比较:两者数据集上表现相似,但 CytoTRACE 总体准确率更高(中位数分别为 74% 和 54%;图 S13C)。推测其可能是由于 RNA velocity模型假设的 mRNA 半衰期和发育时间尺度较短。
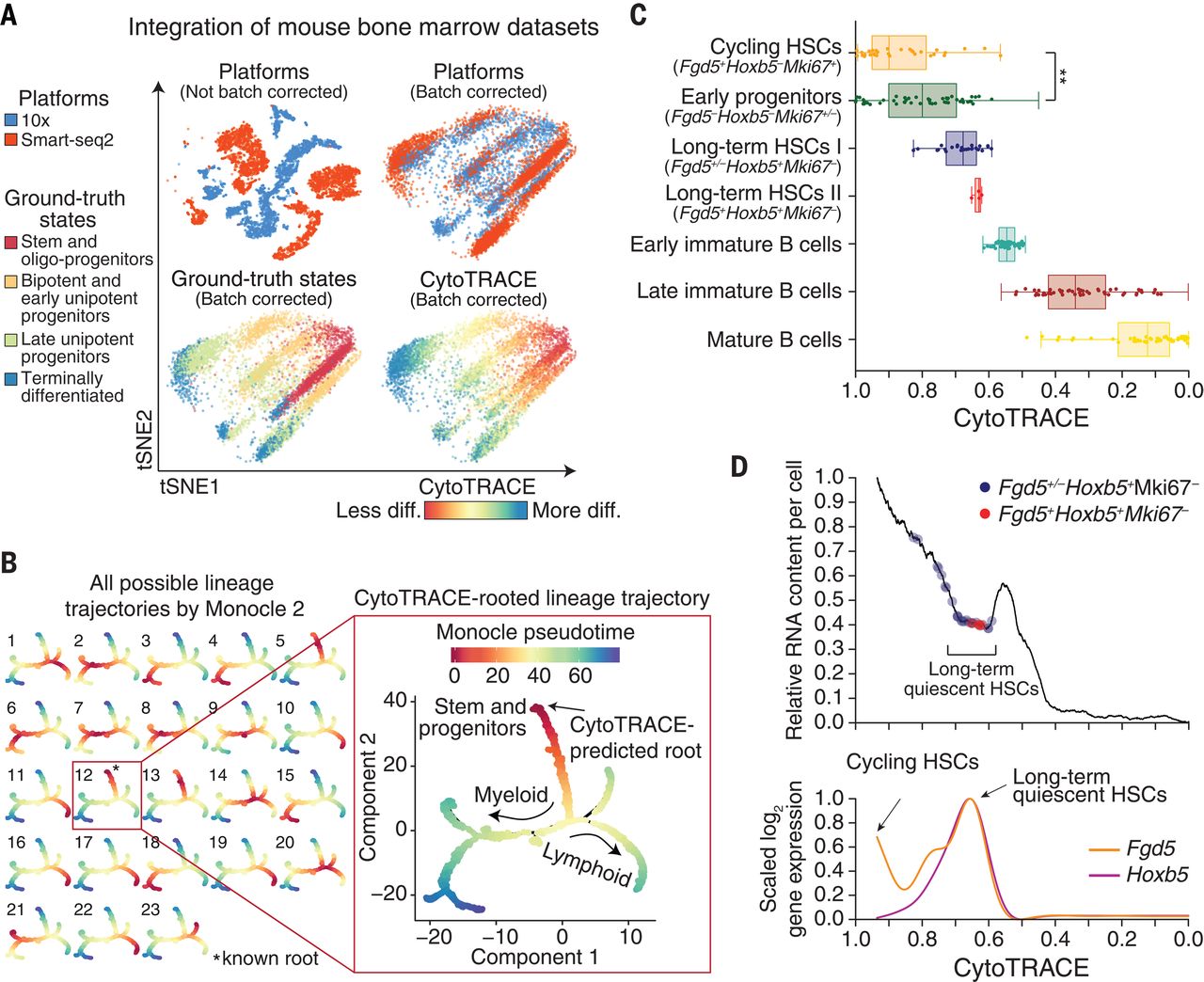
- CytoTRACE 是否可以应用于做过去批次的scRNA-seq 数据集:作者利用Scanorama中的最近邻和高斯核归一化技术合并了几个数据集。整合不同 scRNA-seq 平台上的数据集(图 3A)还是包含发育上不同的细胞类型的数据集(图 S14),Cytotrace的结果都是准确的。
- 寻找干细胞相关基因和谱系关系
- 无需先验知识的情况下识别不成熟表型marker的潜力的验证:见补充文本
- 与Monocle2的比较:当应用于 4442 个骨髓细胞时,Monocle 2 识别出 23 个可能的“root”,从中计算伪时间值(图 3B,左)。相比之下,CytoTRACE 无需用户输入即可轻松识别正确的root(图 3B,右侧,以及图 S16,A 和 B)。这些方法的整合促进了粒细胞、单核细胞和 B 细胞分化过程中谱系特异性调节因子和marker gene的自动识别(图 S16C)。小鼠肠细胞也获得了类似的结果(图 S16,D 至 F)
- CytoTRACE 可以区分循环干细胞和长期或静态干细胞与其下游祖细胞:尽管循环和静止造血干细胞 (HSC) 亚群被正确预测为分化程度较低(low diff),但只有增殖性HSC 的排名显着高于早期祖细胞(图 3C)。通过可视化推断的RNA含量与CytoTRACE的函数(图3D,顶部),我们观察到RNA丰度的存在明显的低谷, 且其CytoTRACE的值与Hoxb5的表达升高相吻合,Hoxb5是长期或静止hsc的标记物(图3D,底部)。由于这些细胞不能单独通过基因计数或RNA含量来鉴定,因此该分析证实了CytoTRACE的实用性,并展示了一种从scRNA-seq数据中阐明组织特异性干细胞的方法。
- 在肿瘤疾病中的应用
- CytoTRACE可用于研究在乳腺癌中未成熟的**管腔祖细胞(luminal progenitor,LP)**及其相关基因:使用 Smart-seq2 方案与荧光激活细胞分选 (FACS) 相结合,我们对来自三个主要人类上皮亚群的细胞进行索引分选和测序。
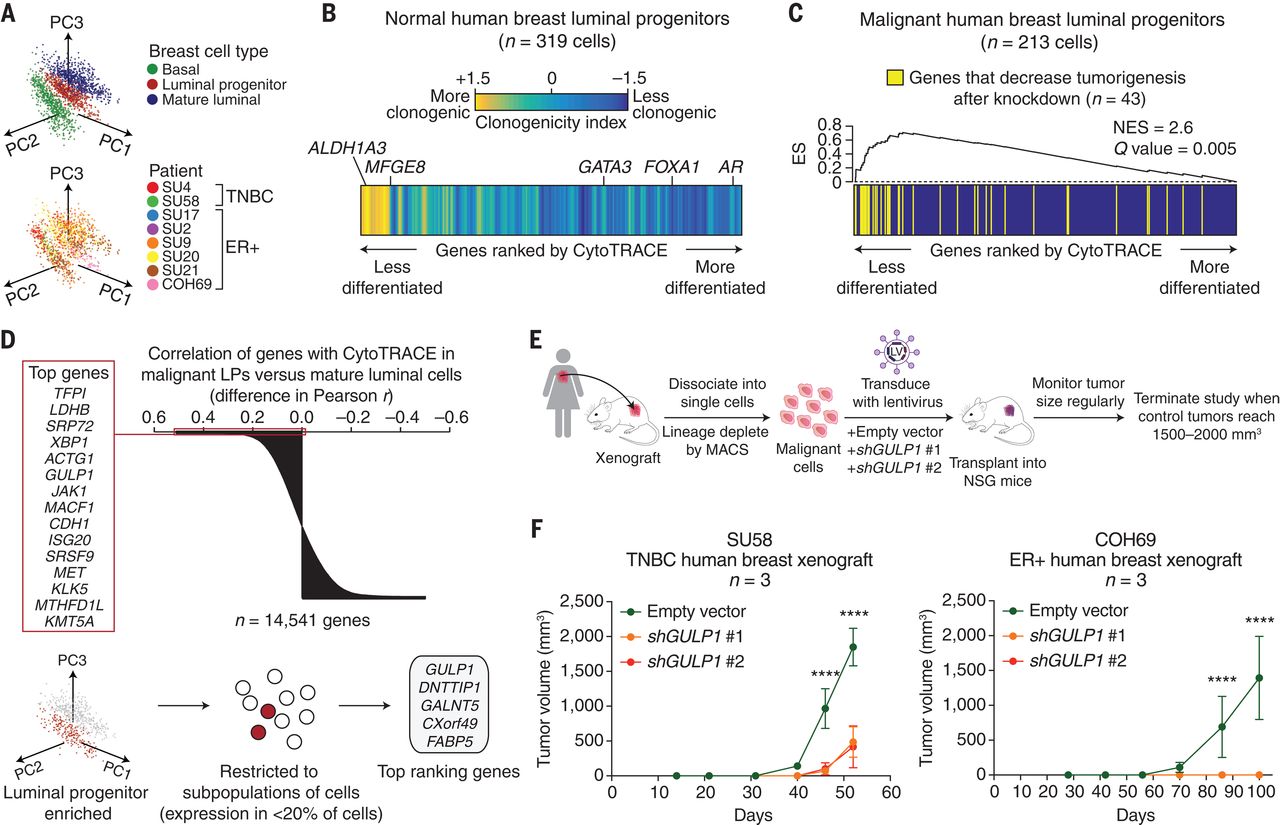
- 验证 CytoTRACE 定义 LP 分化状态的能力:低分化正常 LP [ALDH1A3 和 MFGE8 (39)] 和高分化正常 LP [GATA3、FOXA1 和 AR (39, 40)] 的标记基因通过这种方法成功富集(图 4B) )。此外,在高克隆性正常 LP 中上调的基因 (39) 偏向于预测标记分化程度较低的细胞的基因(图 4B)
- 鉴定与肿瘤发生相关的 LP 基因: 使用Pearson correlation with CytoTRACE对在恶性LPs中表达的基因进行排序,在排序结果中观察到在患者来源的三阴性乳腺癌(TNBC)异种移植(PDX)模型中,通过RNA干扰(RNAi)敲低导致肿瘤细胞活力降低的基因显著富集(图4C)。此外,当我们应用CytoTRACE将肿瘤LPs中的基因与肿瘤**成熟管腔细胞(mature luminal,MLs,它处于LPs的发育下游)**进行优先排序时(39),排名前15位的基因包括乳腺癌中已知的致瘤途径成员[例如MET和JAK1(42,43)],以及未知的候选基因(例如GULP1)(图4D,顶部)。图4E和F用实验证明GULP1 敲低要么抑制肿瘤生长,要么完全消除肿瘤生长。
Conclusion:
- 证明了不太成熟的细胞保持较松散的染色质,以允许更广泛的转录组采样,而分化程度较高的细胞通常会限制染色质的可及性和转录多样性,因为它们specialize。
- 证明每个细胞表达基因的数量是发育潜力的标志。通过利用 scRNA-seq 数据的这一特性,开发了一个解决单细胞分化谱系关系的通用框架。我们设想我们的方法将补充现有的 scRNA-seq 分析策略,对整个多细胞生命中复杂组织中未成熟细胞及其发育轨迹的识别具有影响。
Notes(Optional):
Reference(Optional)
CytoTRACE文献阅读
https://xuan-98-l.github.io/2023/12/22/CytoTRACE/