免疫组(RepSeq)
10X免疫组知识补充
基础知识
免疫组库测序
RepSeq: Immune repertoire sequencing
免疫组库(Immune Repertoire,IR)是指某个个体在任何特定时间点其循环系统中所有功能多样性B淋巴细胞和T淋巴细胞的总和。
T细胞和B细胞分别介导机体的细胞免疫和体液免疫应答,分别通过其表面的T细胞受体(TCR)和B细胞受体(BCR)来识别和结合抗原,进而发挥功能清除病原体或体内肿瘤细胞。TCR/BCR 上存在一块区域叫互补决定区(Complementary Determining Region, CDR),包含CDR1、CDR2、CDR3,其中CDR3最高变,在抗原识别中起关键作用。CDR3由V、D、J三个基因编码。
BCR
BCR 是在 B 细胞膜上表达的免疫球蛋白 (Ig) 的膜结合形式,由两条相同的重链 (IgH:µ,α,γ,δ,ε)和两条相同的轻链 (IgL:κ,λ) 通过二硫键连接而成。BCR 重链分为可变区(V 区)、恒定区(C 区)、跨膜区及胞质区;轻链则只有 V 区和 C 区。V 区由 3 个互补决定区(CDR1、CDR2 和 CDR3)组成,CDR 的氨基酸/基因组成和排列顺序呈现高度多样性,在同一个体内,这种多样性可达 109~1012,构成容量巨大的 BCR 库。正是这种多样性的存在,正常机体几乎能够对入侵的所有异物产生免疫应答。
TCR
TCR 是一种异二聚体,由 T 细胞膜表面两个二硫键连接的跨膜蛋白组成:一个 α 链和一个 β 链,或者一个 γ 链和一个 δ 链。α 和 β 链的 V 区包含 3 个互补决定区 CDRs 和 4 个框架区 FRs,其中 CDR 区是细胞的高变区,与抗原特异性结合,CDR1、CDR2 由 V 基因编码,而 CDR3 由 VDJ 三个基因编码,在免疫细胞成熟过程中 VDJ 基因发生重排从而形成了 TCR 多变性。而且多样性可达 1015~1018。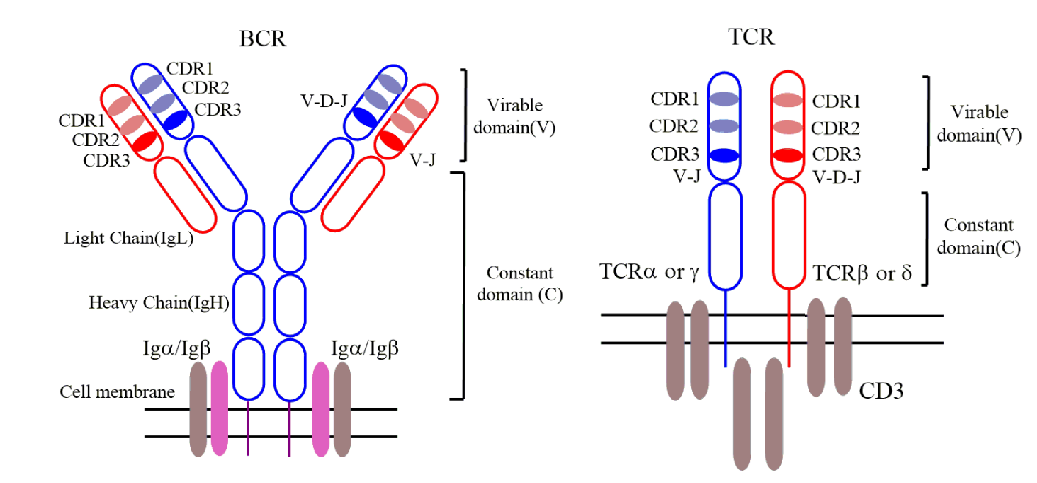
V(D)J 重排
如下图所示,编码重链基因的的染色体包括4种DNA元件(基因片段),分别是 V,D,J,C。
- BCR
在人类B细胞中,V 片段大约有50种, D 片段大约有20种, J 片段大约有6种, C 片段大约有10种。而每个特异 B 细胞就是通过选择每类片段中的一种,并将它们组合在一起,以组装成一个成熟的重链基因。同理, 轻链包括3种DNA元件,分别是 V,J,C。通过这样的方式, B细胞就可以产生种类巨多的抗体分子。但这样还不够,当基因片段组合在一起后,还会有额外的DNA碱基插入或者去除,以增加组合多样性。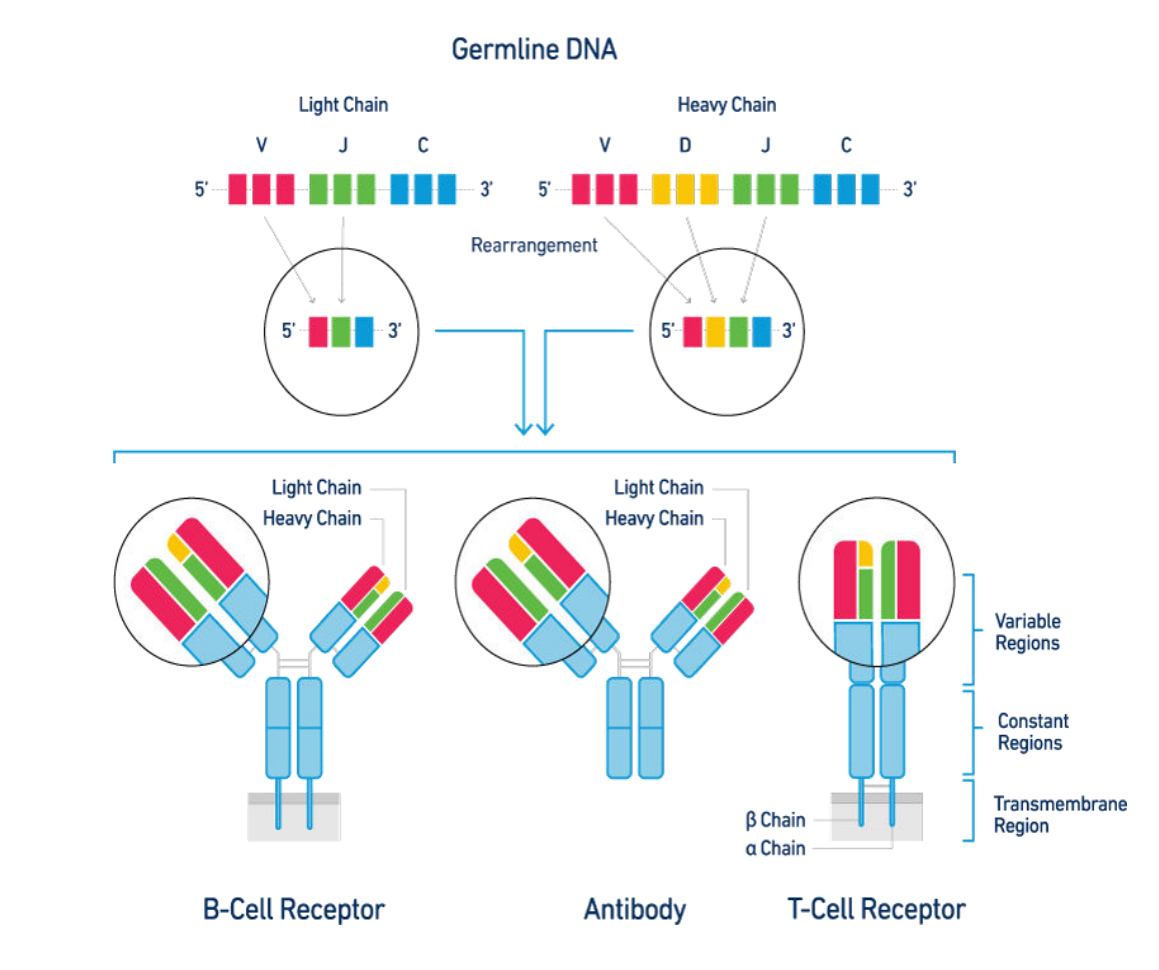
一个B细胞克隆只表达一种BCR,只分泌一种抗体,原因:
- 等位排斥:B细胞一条染色体的重链基因重排成功后,会抑制同源染色体上重链基因的重排;
- 同种排斥:kappa轻链基因重排成功后会抑制lambda轻链基因的重排;
- B细胞中枢免疫耐受的形成——阴性选择
前B细胞在骨髓中发育至未成熟B细胞后,期表面只表达完整的mIgM,此时若mIgM与自身抗原结合,会面临三种命运:
- 细胞凋亡,克隆清除;
- 通过受体编辑改变其BCR的特异性;
- mIgM表达下调,仍然能进入外周免疫器官,但对抗原刺激不发生应答,称为“失能”;
TCR异二聚体的α和β链由可变区(V)和恒定区(C)组成,它们在胸腺发育过程中被拼接在一起,从而产生由每种T表达的单一类型的功能性TCRαβ复合物细胞
β链的V区由可变(V),多样性(D)和连接(J)基因片段编码,而α链的V区由V和J基因片段编码
TRB的候选基因片段数量:
TRBV:52
TRBD:2
TRBJ:13
TRA的候选基因片段数量:
TRAV:70
TRAJ:61
AIRR社区
The Adaptive Immune Receptor Repertoire (AIRR)
- 获取、分析、管理和比较/共享 AIRR-seq 数据集;
- 开发标准化资源,以促进AIRR-seq数据集和分析工具的比较
- 审查 AIRR-seq 数据分析工具是否符合 AIRR;
- 将AIRR-seq数据集与其他“大数据”集相关联,例如微阵列,流式细胞术和MiSeq以及单细胞基因表达数据;
- 涉及使用和共享来自人类来源的AIRR-seq数据集的法律和道德问题。
- 建立将存储AIRR数据的存储库的要求
10x vdj测序
10x Genomics 5’ Chromium Next GEM 单细胞免疫分析解决方案能够以单细胞分辨率同时分析同一组细胞的这些文库:
- T细胞的V(D)J转录组和克隆型
- B细胞的V(D)J转录组和克隆型
- 5’ 单细胞基因表达
- 细胞表面蛋白(抗体捕获)
- 基于条形码的抗原特异性克隆型图谱(Barcode Enabled Antigen Mapping,BEAM) (抗原捕获)
cellranger vdj
cellranger vdj 从 cellranger mkfastq V(D)J 库中或 bcl2fastq 为 V(D)J 库获取 FASTQ 文件,并执行序列组装和成对克隆型调用。它使用Chromium cellular barcodes 和UMI来组装每个细胞的V(D)J转录本。克隆型和CDR3序列作为 .vloupe 文件输出,可以加载到Loupe V(D)J browser中。
分析结果解读:
1 | |
vdj_contig_info.pb: 以 protobuf 二进制文件格式存储contig注释、V(D)J 引用和其他metadata。运行 cellranger aggr 管道所需的文件。此文件仅用于用作 Cell Ranger aggr 管道的输入,不得用作数据源。vdj_contig_info.pb 中包含的信息也可以在其他易于解析的输出文件中找到,例如 all_contig_annotations.json 、 vdj_reference 和 metrics_summary.csv 。
clonotypes.csv: 每个克隆型(clonotype)的高级描述。包含clonotype_id(按照frequency排序,ID从低到高);Frequency(观察到的具有此克隆型的细胞barcodes的数量);Proportion(观察到的具有此克隆型的细胞barcodes的比例); cdr3s_aa(CDR3氨基酸序列,链包含TRA,TRB,TRG,TRD,IGK,IGL或IGH);cdr3s_nt(CDR3核苷酸序列);inkt_evidence(对于T细胞,这列指示clonotype是否是一组 invariant natural killer T ,即inkt细胞);mait_evidence(对于T细胞,这列指示是否为mucosal-associated invariant T,即MAIT细胞)
注:关于INKT及MAIT细胞的信息见链接
- consensus_annotations.csv:每个克隆型共有序列(consensus sequence)的高级和详细注释。
注:10X的Csv表格输出都是用于高级注释,每行有一个contig, consensus, 或者 clonotype.
all_contig_annotations.csv:CSV 格式的所有重叠群(contigs)(来自细胞和背景条形码)的高级和详细注释。
filtered_contig_annotations.csv:来自细胞相关barcodes的每个高置信度重叠群的高级注释。这是一个all_contig_annotations.csv 子集.
all_contig_annotations.bed:BED 格式的所有重叠群(来自细胞和背景条形码)的高级和详细注释。Browser Extensible Data,字面意思是“(基因组)浏览器可延展数据”,是一种基因组学中用于表示、标记基因组区域位置信息中的文件格式,与IGV等可视化工具一同使用。
all_contig_annotations.json:JSON 格式的所有重叠群(来自细胞和背景条形码)的高级和详细注释。Json格式含有详细的注释,包括contig的完整序列,contig的具体坐标和氨基酸翻译序列。
airr_rearrangement.tsv : AIRR格式的带注释的重叠群和 V(D)J 重排的共有序列。
vloupe.vloupe: Loupe V(D)J Browser readable file
典型V(D)J转录的结构![]()
UTR:未翻译的区域;
FWR:框架区域(Framework region);
CDR:互补性决定区域
名词解释:
contig:带有相同的 cell barcode 和 UMI 且比对到相同转录本序列的 reads 被组装为 contigs。(由组装产生的连续的碱基序列)
full-length: 如果重叠群与 V 基因的起始部分相匹配,继续下去,并最终与 J 基因的末端部分相匹配,则该contig是全长的。(可以没有d_gene)
Clonotype(克隆型): 一组适应性免疫细胞,是完全重组、未突变的共同祖先的克隆后代。T细胞克隆型通常由重排的TCR的核苷酸序列来区分,在大多数脊椎动物物种中,TCR不经历体细胞超突变(SHM)。如上所述,B细胞克隆型通常在核苷酸水平上彼此不同。由于这个原因,B细胞克隆型也经常包含多个精确的亚克隆型.
10X的判断方式:
优先考虑TRA和TRB所有cdr3_nt都匹配,分配到同一个clonotype。(所有的Exact subclonotype都匹配)
若A细胞中只含有单一的TRA序列(该细胞不含TRB),该序列完全匹配到B细胞的TRA(B细胞可以含有TRA或TRB),则会为A细胞的TRA序列分配一个clonotype ID,且把B细胞全部分配到该Clonotype ID上.(A细胞中只有一个Exact subclonotype,且该Exact subclonotype与B细胞的Exact subclonotype匹配,即使B细胞存在其余Exact subclonotype,A和B细胞也会被分到同样的Clonotype ID)
Exact subclonotype(精确亚克隆型): 在核苷酸水平上具有相同免疫受体序列的克隆型细胞的一个亚群,横跨整个V、D和J基因以及V(D)J连接。精确的亚克隆型共享相同的V、D、J和C基因注释(例如,具有相同的V(D)J序列但不同的C基因或同型的细胞被分成不同的精确亚克隆型)。
Consensus(一致性序列):给定clonotype ID的一致序列是该链在第一个精确亚克隆型的核苷酸序列。
productive(生产性):
如果满足以下条件,则重叠群被称为生产性的(有prodictive 的TCR/BCR才能翻译有功能的蛋白):
- full length: contig 与 V 基因的初始部分相匹配。contig 继续,最终匹配 J 基因的末端部分。
- 可编码正常氨基酸:V片段的起始部分与config的起始密码子相匹配(在 10x Genomics 提供的人类和小鼠参考序列中,每个 V 区段都以起始密码子开头。),也就是有起始密码子;V 片段起始和 J结尾之间没有终止密码子;J 停止减去 V 开始等于一模三。这只是说 V 和 J 片段上的密码子在框内(编码范围内没有移码突变)。
- CDR3:有一个带注释的 CDR3 序列。
- 结构:令 VJ 表示 V 和 J 段的长度之和。让 len 表示 J 停止减去 V 开始,在重叠群上测量。那么 VJ -len 介于 -25 和 +25 之间,除了 IGH,它必须介于 -55 和 +25 之间。施加此条件是为了排除不太可能与功能蛋白相对应的异常结构变化。
- high_confidence contig(高质量contig): 有两个以上的UMI支持的productive contig为高质量的contig.
cellranger multi
cellranger multi 从单个 GEM 孔中获取 5’ 基因表达、基因barcodes(抗体捕获、CRISPR Guide Capture和/或抗原捕获)和 V(D)J 文库的任意组合的 FASTQ 文件 cellranger mkfastq bcl2fastq 。它对基因表达和/或Feature Barcode (FB)执行比对、过滤、barcodes计数和 UMI 计数。它还对 V(D)J 文库执行序列组装和成对克隆型调用。此外,基因表达数据提供的cell call用于改进V(D)J文库推断的cell call。
分析结果解读:

该 cellranger multi pipeline通过丢弃在相应的 5’ 基因表达数据集中未调用的任何细胞来改进 V(D)J 数据集中的细胞调用。通过将 V(D)J 结果中调用但未在 5’ 基因表达结果中调用的细胞指定为 V(D)J 数据中的背景 GEM, cellranger multi 可以减轻 V(D)J 数据中可能出现的任何超调问题。这种改进的细胞调用只有在从同一样品中同时测序5’基因表达和V(D)J文库时才有可能。
如下图所示,最终的 V(D)J 单元调用(交集区域)排除仅由 vdj 管道调用的单元(黄色区域)。(最终获得的细胞barcodes在VDJ数据和转录组数据中是一致的)
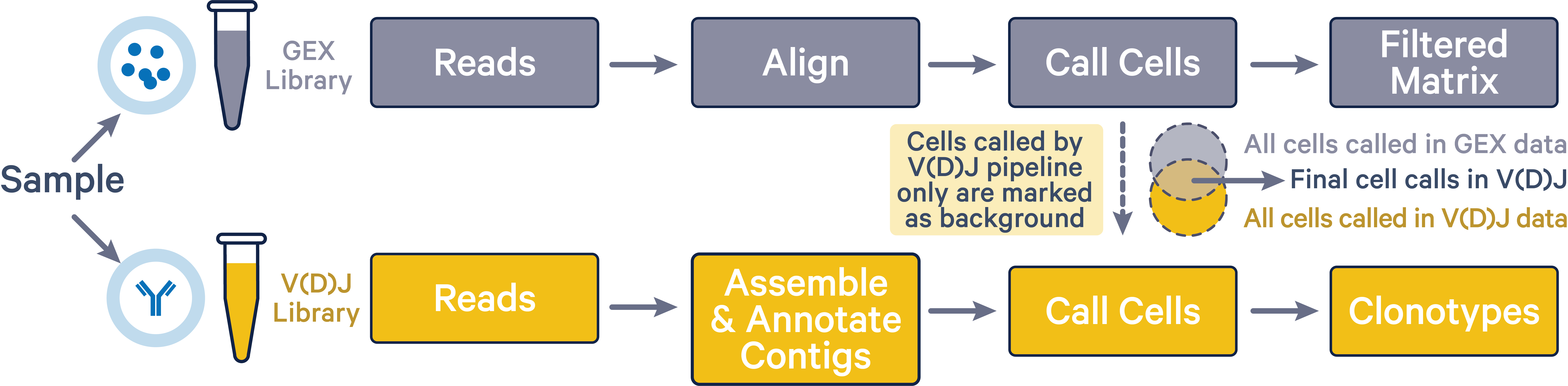
5’ 基因表达细胞调用不受 cellranger multi 管道影响。基因表达文库代表了每个GEM中捕获的整个聚腺苷酸化mRNA转录本库。然后选择性扩增基因表达文库中的VDJ-T或VDJ-B转录本以创建V(D)J文库。因此,与 V(D)J 文库相比,基因表达文库具有更强的检测含有细胞的 GEM 的能力。如果 cellranger multi 管道同时使用 5’ 基因表达和 V(D)J 数据运行,则从 V(D)J 细胞集中删除在 5’ 基因表达数据中未称为细胞的条形码。
从Cell Ranger 7.0开始,multi可以自动估计或指定 expect-cells 预期的细胞数量(例如,复制先前的分析)(force cell)
该 cellranger multi 管道允许用户分析富集为 gamma (TRG) 和 delta (TRD) 链的 TCR 库。但是,Gamma-delta 分析不是受支持的工作流程,并且无法保证算法性能。TRG/D 输出位于 和 outs/per_sample_outs/mySample/vdj_t_gd 文件夹中 outs/multi/vdj_t_gd 。文件夹中的 vdj_t_gd 输出文件与 的 vdj_t/b 输出文件类似。
cellranger aggr
Cellranger AGGR 整合来自多次运行 cellranger count cellranger vdj 、 或 cellranger multi 的输出。该管道将 aggr 单个基因表达和Feature Barcode (FB)运行规范化到相同的测序深度,重新计算feature barcodes矩阵并对组合数据进行分析。该pipeline还会重新计算组合数据的 V(D)J 克隆型分组。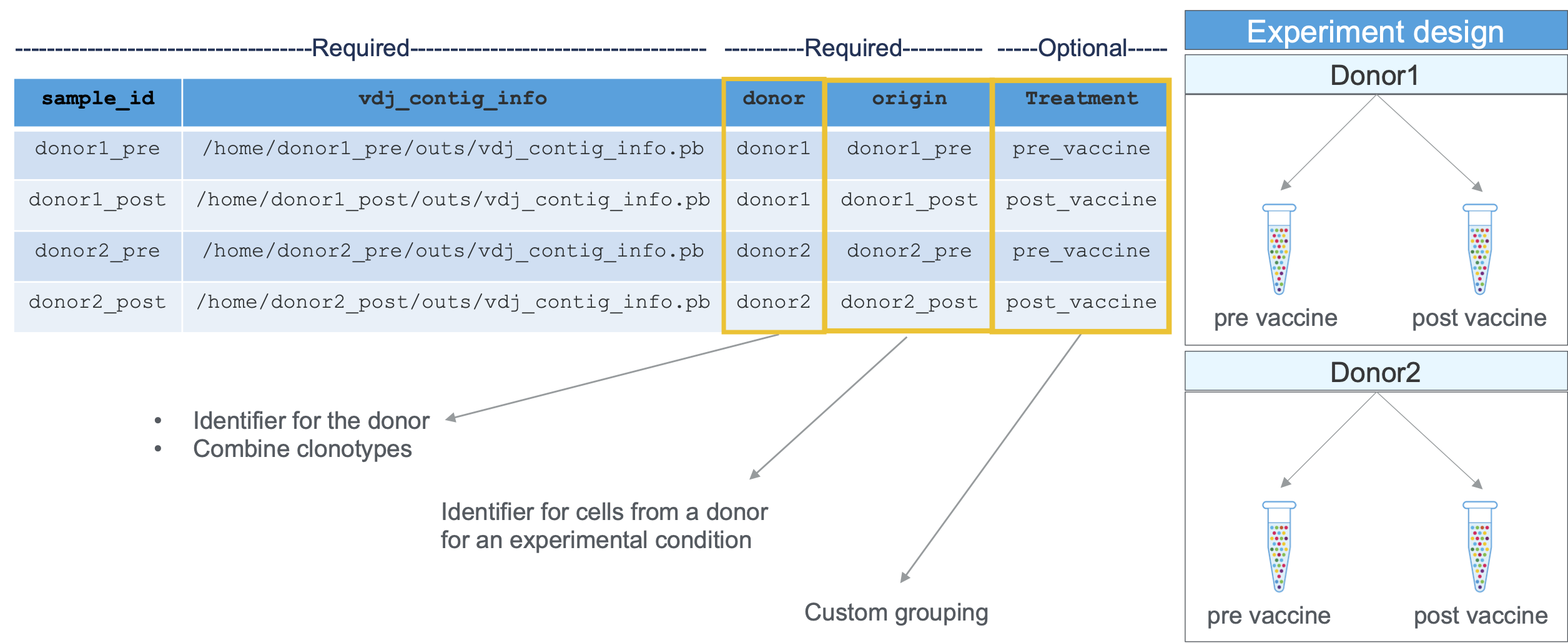
Cellranger VDJ算法概述
cellranger vdj 针对FASTQ 文件执行序列组装和配对克隆型调用。该管道使用10x barcodes和 UMI 来组装每个细胞的 V(D)J 转录本。克隆型和CDR3序列作为 .vloupe 文件输出,可以加载到Loupe V(D)J浏览器中。
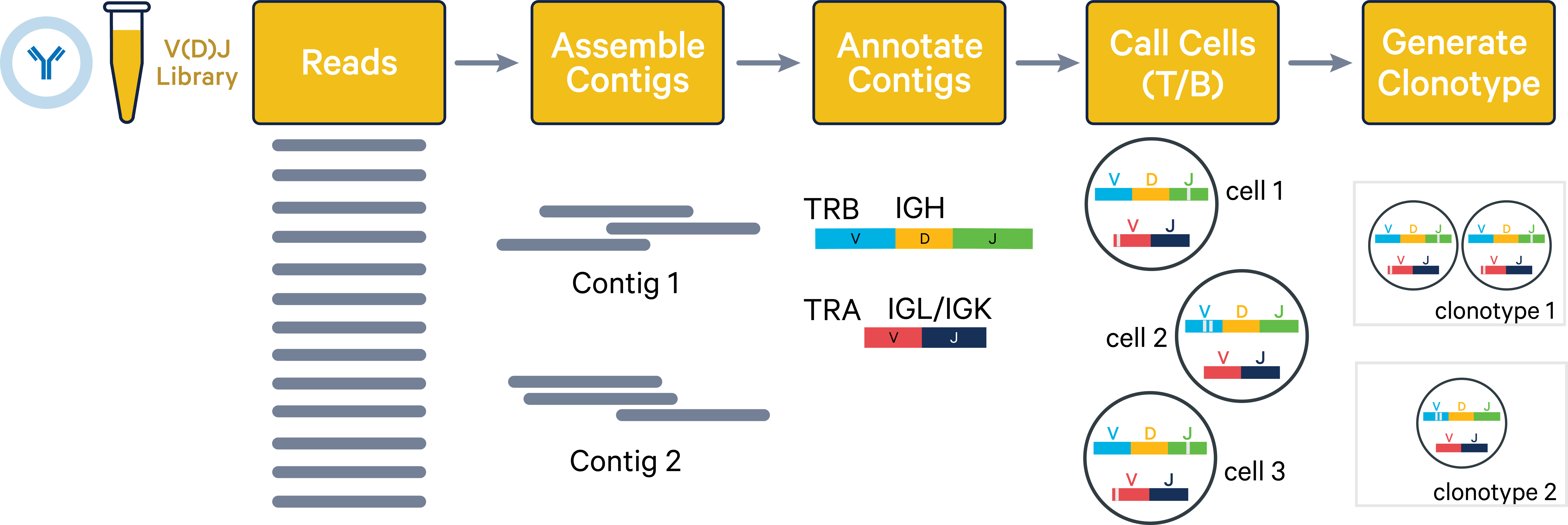
- contigs组装
- 过滤掉可能由PCR错误、测序错误等引起的带有背景barcodes和UMI的读取。
- 一些细胞具有极高的覆盖率。高覆盖率可能是由于真正的高测序覆盖率,或浆细胞中的高mRNA表达(常见于BCR)。转录本的超高覆盖率(大于 80,000 个reads)会降低计算性能并添加很少的额外信息。因此,每个barcodes的覆盖范围上限为最多 80,000 次reads。如果任何给定的 10x barcodes的读取次数超过 80,000 次,则对读取进行downsample。
- 从reads的 5’ 和 3’ 端修剪adaptor和引物序列。
- 从所有观察到的 GEM/条形码中的每条链生成全长转录本(contigs)。
- 注释Contig
V(D)J重叠群注释的三个目标是:
1)定义V,D和J片段与contig的比对
2)识别CDR3序列
3)从这些数据中确定contig是否productive,这意味着它可能对应于功能性T细胞或B细胞受体。
- 用 V(D)J 片段标签注释contig,并定位形成转录本的 CDR3 区域。
- 过滤全长且productive的重叠群。
- 重叠群置信度:不正确的克隆型可能来自细胞外mRNA或双峰等来源。为了防止这种情况,将评估重叠群的置信度,并排除那些被宣布为低置信度的重叠群。所有非生产性重叠群均被声明为低置信度。对于参考辅助装配,如果满足以下任一条件,则生产重叠群的置信度较低:有四个以上的productive contigs;每个链有两个以上的重叠群(例如三个TRA重叠群):预计每个细胞相关条形码都有一个productive TRA chain和一个productive TRB chain用于T细胞,一个productive重链和一个productive轻链用于B细胞。额外productive contigs不太合理;少于三个filtered UMI (每个 UMI 至少具有三个reads);所有productive contig最多有一个连接 UMI 支持,并且每个过滤的 UMI 少于 5 个,每个 UMI 至少具有三个reads,或者有两个以上的生产性重叠群;
细胞识别
识别包含 T 或 B 细胞的barcodes/GEMs。生成clonotype(克隆型)
将细胞相关barcodes分配到克隆型中并过滤掉一些细胞。
主要从all_contig_annotations.json的Cell Ranger文件中获取其信息。
在克隆性计算过程中,会对细胞进行过滤(过滤Technical artifacts)。
T细胞:T细胞受体(TCR)中缺乏体细胞超突变(SHM)会产生具有相同V(D)J 转录本的生物克隆型。技术伪影(Technical artifacts )(例如在逆转录中出现)可能导致计算出的克隆型具有独立的差异。这些很少见。
B cells: Fully rearranged B cell receptors (BCRs) can undergo SHM, which can increase antigen affinity. Thus for BCRs, V(D)J transcripts in a clonotype can differ at any position, as shown below:
B细胞:完全重排的B细胞受体(BCR)可以经历SHM,这可以增加抗原亲和力。因此,对于BCR,克隆型中的V(D)J转录本在任何位置都可能不同,如下所示: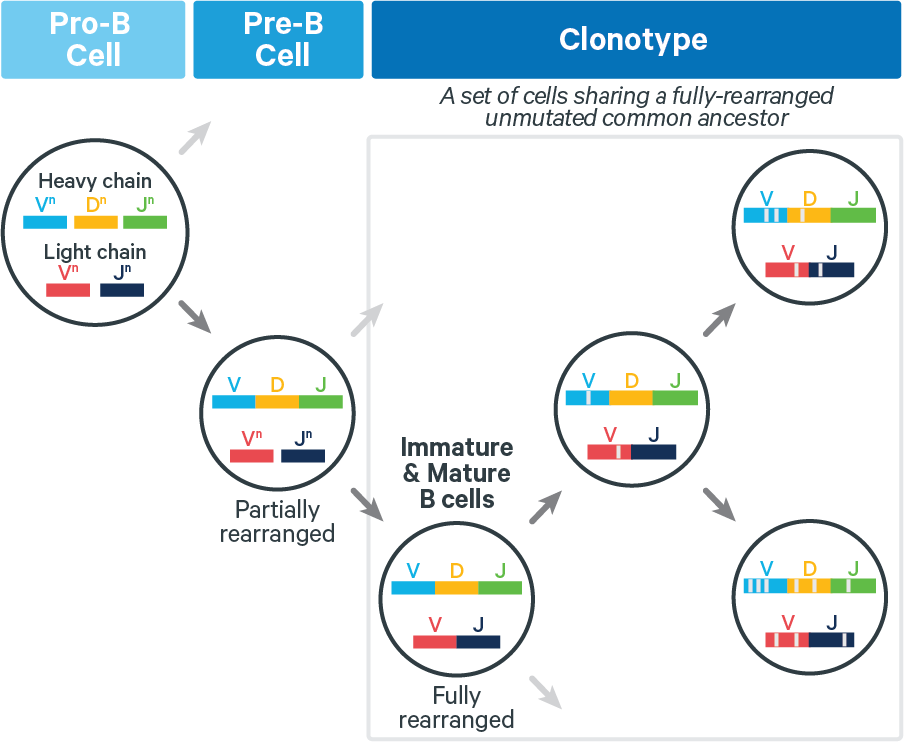
Cellranger只考虑V 基因位置的种系变异评估(SHM), 不考虑J基因的SHM。
步骤:
(1) Exact subclonotype grouping精确的亚克隆型分组
如果细胞具有相同的V(D)J转录本,则将其放入称为精确亚克隆型的组中。仅使用productive重叠群。精确亚克隆型必须具有相同数量的链。它们的V(D)J序列和恒定区域基因分配也必须相同。精确亚克隆型不需要具有相同的 5’ UTR。此外,该算法不会在 5’ UTR 或恒定区域中测试 SHM。
(2)将精确亚克隆型连接成克隆型
基于将每对精确亚克隆型相互比较而迭代合并为克隆型。具有共同差异和最小CDR3突变的两个细胞被认为是同一克隆型。此处简要介绍了合并条件。
10X enclone 算法摘要
10X enclone 算法文献
10X enclone 命令行版本(Beta版本)
相关文献
Cellranger中默认启动如下Enclone配置
Cellranger术语表
Web summary metrics for V(D)J Troubleshooting
10X官方推荐下游分析软件