基于SCENIC的转录因子分析原理讲解与实操
基于SCENIC的单细胞转录因子分析原理讲解与实操
转录因子的定义
转录因子, transcription factor(TF), 是调节基因转录的蛋白质,它们通常是由蛋白质,或者短的非编码 RNA 组成。转录因子通常以复合体的形式工作,形成多重相互作用,允许对转录速率进行不同程度的控制。
它们的功能是调节(打开和关闭)基因,以确保基因在细胞和生物体的整个生命过程中在正确的时间以正确的数量在正确的细胞中表达。TF在整个生命过程中协调指导细胞分裂、细胞生长和细胞死亡;指导胚胎发育过程中的细胞迁移和组织发育;或间歇性地响应来自细胞外的信号,例如激素。人类基因组中有多达 2600 个转录因子。
真核生物通常在基因上游有一个启动子区域,或者在基因上游或下游有一个增强子区域,具有被各种类型的 TF 识别的某些特定基序(motif)。转录因子结合、吸引其他转录因子并形成一个复合物,最终促进 RNA 聚合酶的结合,从而开始转录过程。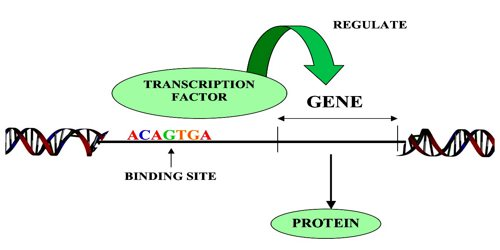
细胞的转录状态来源于潜在的基因调控网络(GRN),它包括一定数量的转录因子(TFs)和辅因子及其下游靶基因。SCENIC可识别TFs与潜在靶基因的共表达模块(Regulon),进而推断基因调控网络及细胞状态。
Tips:Regulon是受同一个TF调控的靶基因的集合。
SCENIC简介
SCENIC开创性的工作是在R中完成的,结果于2017年发表在nature methods上,后来研究人员对其进行了python的实现,开发了pyscenic。
pyscenic的一大优点是使用了基于分布式计算的框架,因此对运行速度有了极大的提升,但由于R版本的SCENIC更方便进行与seurat包的对接,产生比较多的中间文件,方便个性化分析和下游的可视化处理。
若需使用pyscenic, 可以参考natrue protocal中的论述和官方链接进行学习,pyscenic可以使用nextflow或者docker直接进行安装使用,流程也是比较简单。
SCENIC代码实操
0.Pre-processing
由于R版本的SCENIC需要使用GENIE3算法,它基于随机森林,运行时间较长长,因此在demo数据分析时需要对数据集进行降采样的处理,也就是抽出其中的一部分细胞和基因进行分析,以减少内存和cpu消耗(此操作不适合正式分析)
1 | |
1.共表达(Co-expression)
SCENIC正式工作流程的第一步是根据表达数据推断所有转录因子的潜在靶标。为此,我们使用GENIE3或GRNBoost。
输入文件是过滤后的表达矩阵(可以是csv格式 ,loom格式或者Seurat对象里的counts矩阵)。
GENIE3/GRNBoost使用了不同的机器学习算法,GENIE3使用了基于决策树的bagging算法,使用了随机森林整合多个决策树。 GRNBoost使用了boosting 。但两者的输出结果是相似的,都是一个矩阵,包括转录因子,靶标基因和权重(Importance)构成的表格,我们将其称之为共表达模块(moudle)。
图中有N个基因,将会以每个基因作为输出(靶基因表达量),其余基因作为输入(调控基因表达量)来构建N个随机森林模型。进而计算每一个模型中,每个调控基因对靶基因的重要性,从而得出它们之间的调控关系。然后根据所有模型的调控关系进行排序。权重是转录因子对每个基因表达情况的影响的一个程度。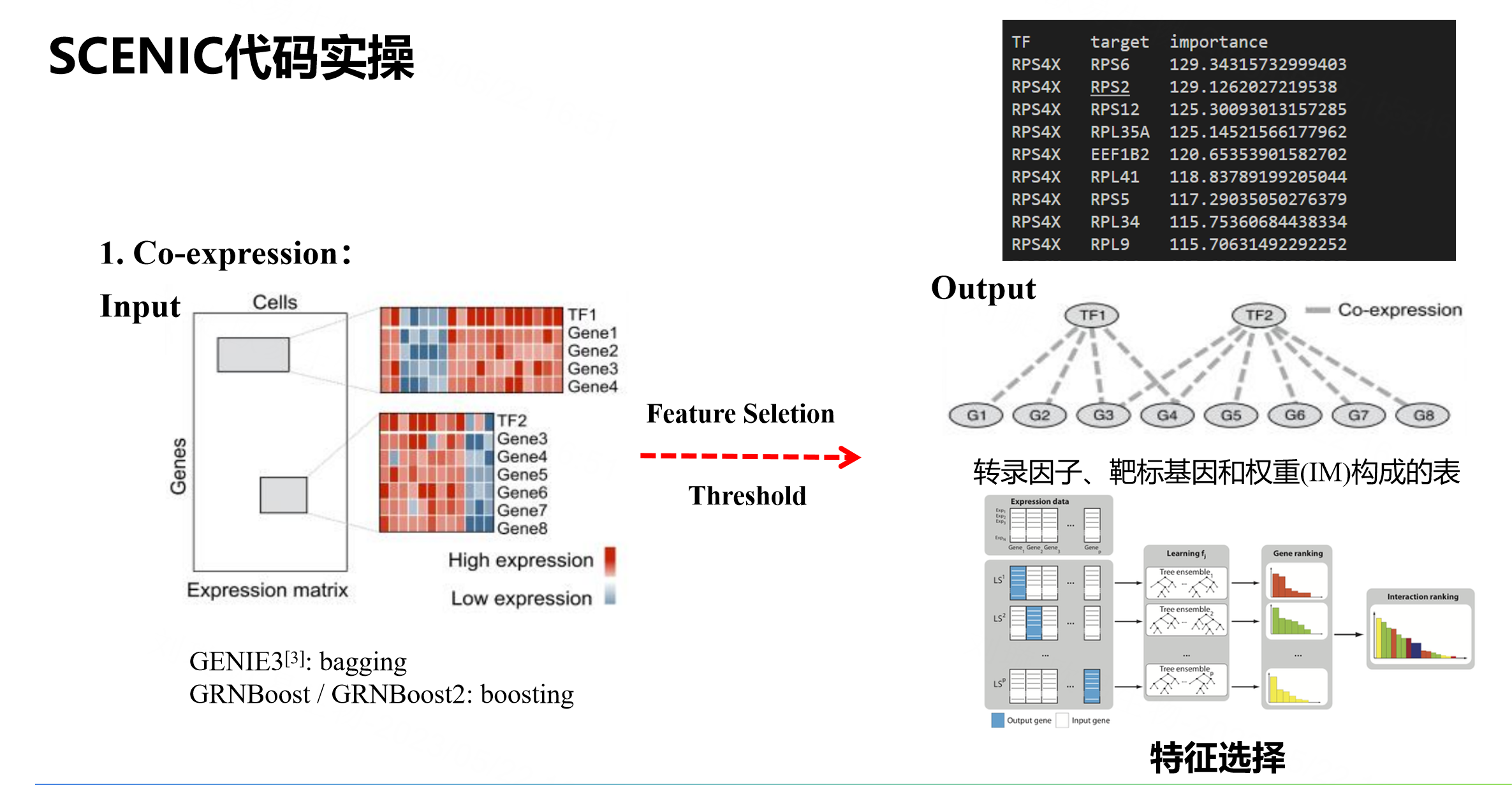
为了在SCENIC的多个步骤中保持一致的设定和输出结果文件的命名,SCENIC包中的大多数函数使用scenicOptions作为公共对象,它存储当前运行的选项并使用默认参数代替大多数函数的“参数”,从而保证了一致性。这一步需要在SCENIC运行之初就使用initializeScenic进行设置,需要设置的内容包括org(也就是物种),dbDir(数据库的路径)等。
1 | |
Tips:
RcisTarget数据库只支持三个物种
- 小鼠 mgi
- 人类 hgnc
- 果蝇 dmel
dbDir下存放四个文件:
- feather文件:保存Gene-motif排名数据库,为每个motif提供所有gene的排名,在接下来的motif富集分析中会用到
- hs_hgnc_tfs.txt: 保存转录因子列表,这个文件在构建共表达矩阵的时候就要用到,因为要告诉SCENIC,你的输入矩阵里哪些基因是转录因子。
- tbl文件:转录因子的Motif注释文件,该文件对每一个motif注释其所对应的TF,这也是motif富集用到。
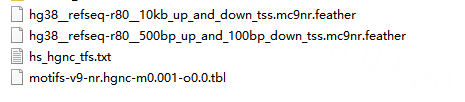
注意下feather羽毛文件的版本,由于在SCENIC在20220816日更新,现在pyscenic仅支持 Feather v2 格式的数据库 ( ctxcore ),它允许使用最新版本的 pyarrow ( ) 而不是旧版文件。我们在使用R版本的Scenic时,由于arrow包无法读入最新版本的feather格式,因此在下载数据库的时候需要下载旧版文件。
接下来我们对基因进行过滤,目的是去除最可能是噪音的基因。
主要是根据两个点对基因进行过滤:该基因的count数,和在多少细胞中被检出。具体的阈值是case by case, 我们这里是使用minCountsPerGene保留在所有样品中至少有1个counts的基因。minSamples设置为保证保留下来的基因能在至少1%的细胞中检测得到。使用geneFiltering函数对基因进行过滤。
1 | |
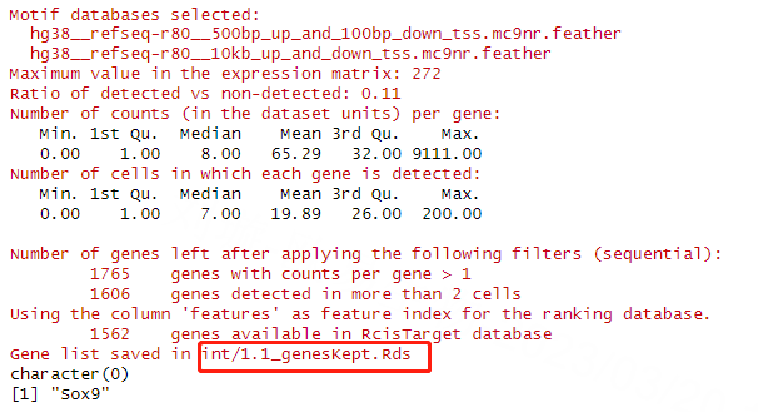
可以看到SCENIC生成的中间文件和图文件将会保存到int文件夹中,并且会按顺序整齐排列好。可以使用这些中间文件来检查之前做过的每个步骤,或者是以一些不同的设置重新运行其中的分析。重要的输出结果会保存到output文件夹中。
这步生成的过滤之后的基因列表就被保存在1.1geneKept.rds中了。使用保留下来的这些基因对矩阵的行进行筛选,最终的这个exprMat_filtered对象所保存的count matrix就是GRN的输入文件。
接下来我们读取转录因子列表文件(在数据库路径下),使用Seurat包的CaseMatch函数,对转录因子名称进行过滤,仅保留匹配到矩阵中的基因名。
1 | |
现在我们正式开始构建共表达网络。R scenic默认的方式是使用GENIE3进行共表达网络的构建。GENIE3使用的是随机森林的方法,他对样本和特征的选取是随机的,所以每次运行的结果可能会稍有不同。因此,我们可以使用Seed随机数种子来保证结果的一致性。GENIE3的运行速度较慢,适合5千细胞以下的数据集。
另一种方法是使用grnboost2进行共表达网络的构建。可以从R中切出去直接运行pyscenic,或者如演示代码所示,使用reticulate R包直接调用Arboreto模块来直接执行grnboost2,由于pyscenic需要配置额外的python环境,我们在这里只使用genie3进行演示。
1 | |
Tips:GENIE3和Grnboost运行结果的差异。
注意,grnboost2和Genie3的输出文件的表头不同,可以看到GENIE3的表头是weight,而Grnboost的表头是importance。为了让scenic识别我们用grnboost生成的文件,需要注意修改列名,变为weight。
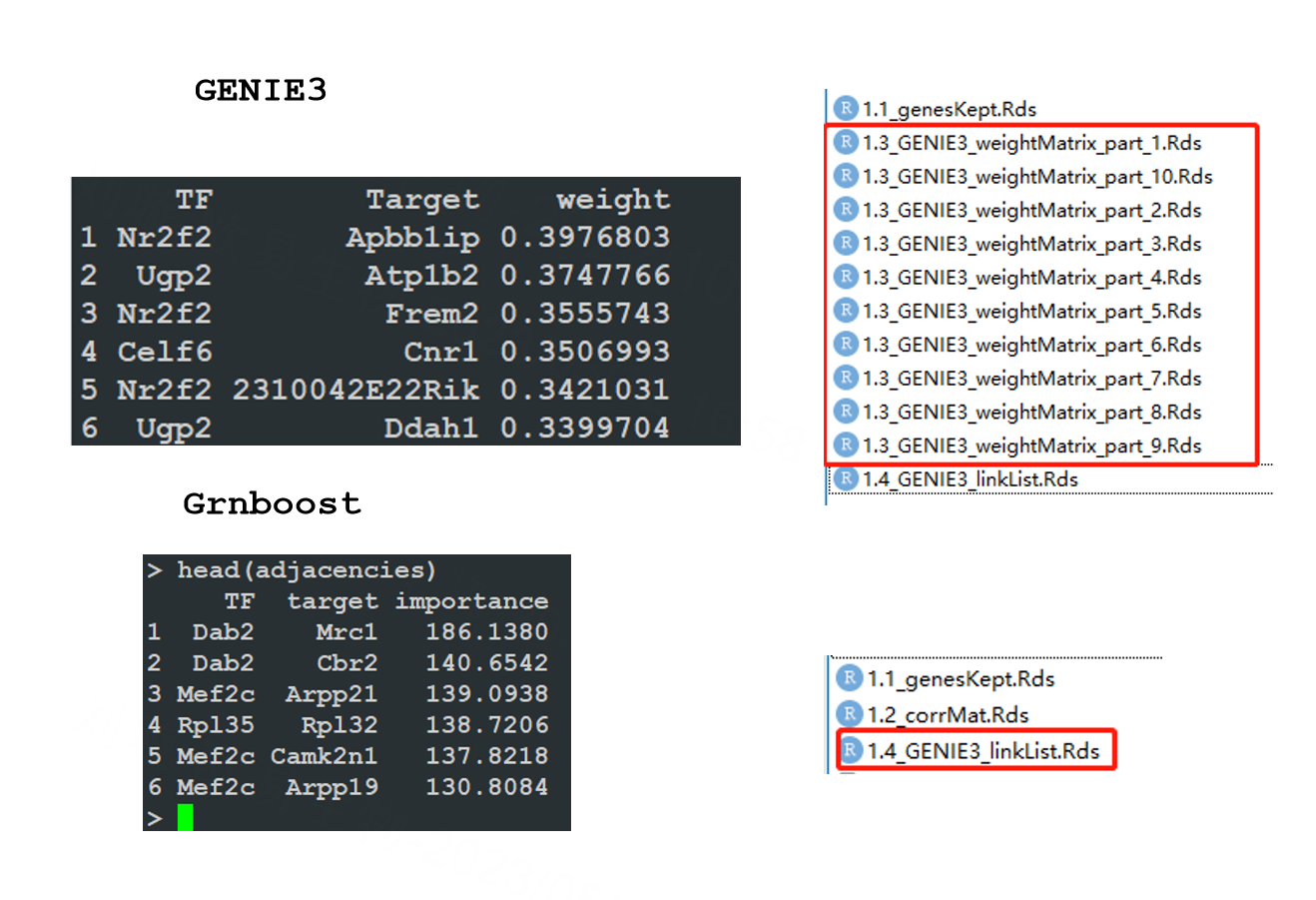
若是使用pyscenic进行共表达分析,后面可视化使用R语言,可以注意下这点区别。 Genie3的输出文件里还有一系列的1.3文件,但都不重要,这一步最重要的是1.4的矩阵。
Tips:查看SCENIC默认的命名规范。左侧提醒我们这个文件里保存了什么信息,右侧是文件名,方便我们查找。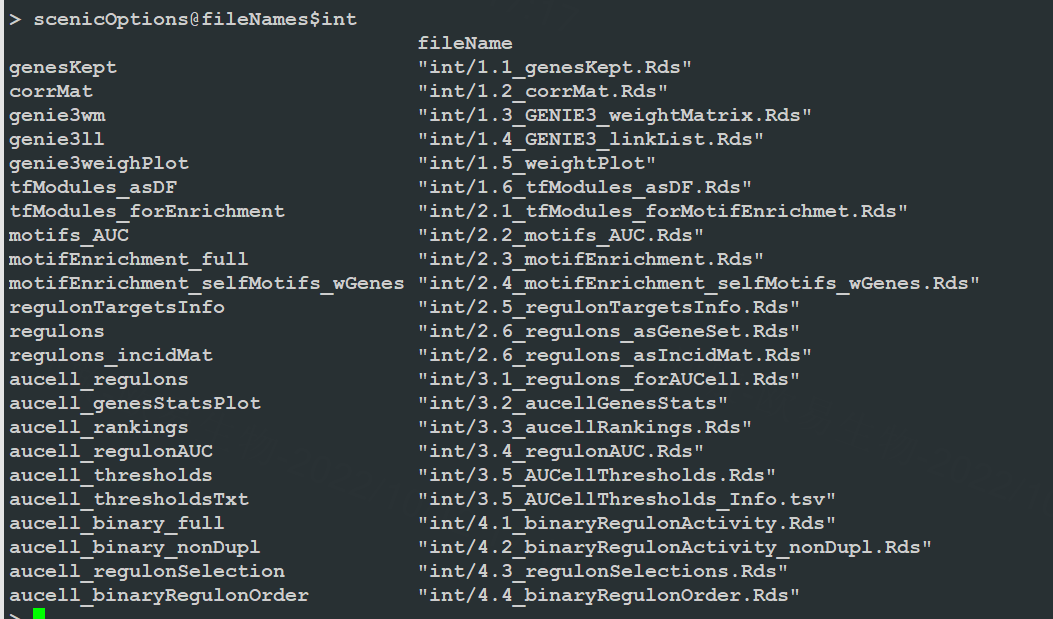
上一步生成的adjacencies的共表达的结果中既有正调控也有负调控,需要相关性矩阵来协助筛选共表达模块中与转录因子正相关的基因。因此我们使用runcoorelation命令,储存基因基因之间spearman相关性的矩阵。
1 | |
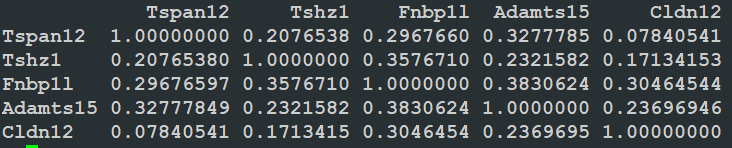
基于这个矩阵,可以使用runSCENIC_1_coExpressNetwork命令推断共表达模块。之前提到过scenicOptions存储了默认的参数,因此可以直接输入scenicOptions以代替coExpressNetwork命令的参数。
1 | |
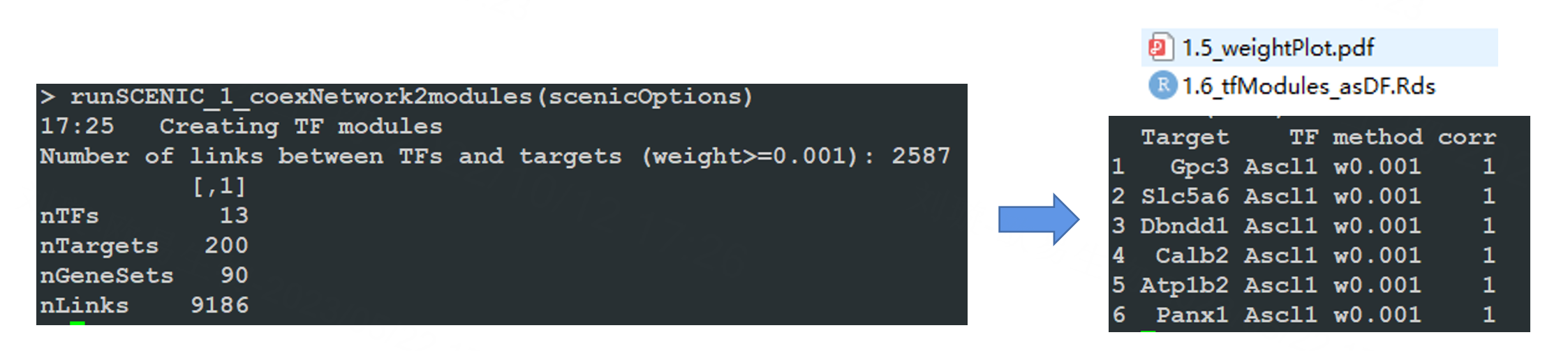
module(模块)现在已经生成了,并且被保存在1.6_tfModules_asDF.Rds文件中。里面显示了转录因子,靶基因和对IM值,就是权重的过滤标准(毕竟module不能无限大嘛)和corr相关性列,scenic只选取数值为1的数据用于后续分析。
查询SCENIC代码参数网址
2. 使用motif验证TF与gene的靶向关系
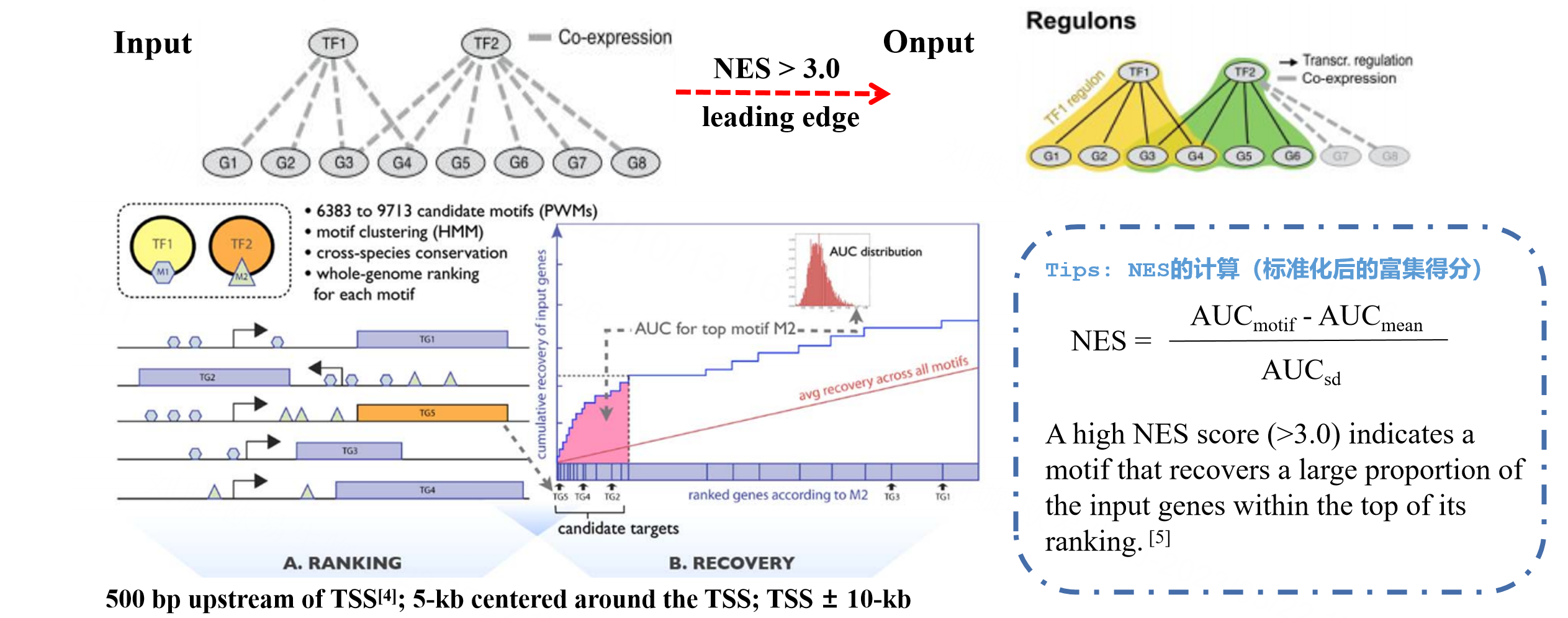
基于表达量矩阵生成module之后,就需要验证TF与gene的靶向关系。TF是通过直接与DNA序列上基因转录起始位点附近的motif序列结合而发挥作用的,因此我们可以通过查看gene上是否存在TF结合的motif序列来去除转录因子不与其直接作用的靶基因。
我们以左下角这个图为例来说明,假设我们有转录因子1,它与motif1进行结合,调控基因转录。还有一个转录因子2,它与motif2进行结合。假设对于某个module,我们有五个靶基因,target gene 1-5,我们根据motif的富集情况开始对基因进行排名。
基于gene-motif数据库,也就是我们输入的feather文件,我们已经获得了每个基因转录起始位点附近的motif富集情况,比如对于TG5来说,它的转录起始位点附近有3个Motif 2,比TG1到4都多,所以我们把它排在第一名。这个module的基因集中,TG4的转录起始位点附近有2个motif,所以把它排在第二名。以此类推,就可以对module里的所有基因进行排名。
然后我们绘制积累曲线,这个图里的横坐标是该物种全基因组的基因对M2富集情况的排名,我们在全基因组排名前5%的基因里寻找我们这个module里有的基因,每找到一个就在y轴上加1。然后计算积累曲线的曲线下面积,就是AUC值。使用公式:该motif的AUC值减去所有motif中的AUC值再除以AUC的标准差,我们就获得了标准化的富集得分。
NES大于3,证明该motif在基因集的大部分基因里出现富集,并且基因排名靠前。由此我们可以剔除非转录因子直接调控的基因,最终获得TF和靶基因的(regulons)。
1 | |
这里我们添加了Coexmethod参数,这个参数是以选择用哪个IM阈值生成的module,不同的IM阈值最终会产生不同大小的基因集。如果不加Coexmethod参数,就默认的六种过滤方法生成的module都用,包括基于固定阈值(0.001和0.005);基于每个转录因子的前50个靶基因产生module;根据每个基因的前5,10,或50个转录因子产生module;

这一步生成的文件主要就是这个转录因子富集结果的网页展示图。数据库直接注释到的和同源基因推断转录因子为高可信度,使用Motif的序列相似性注释到的转录因子为低可信度结果。
Tips: Step2_MotifEnrichment_preview.html文件展示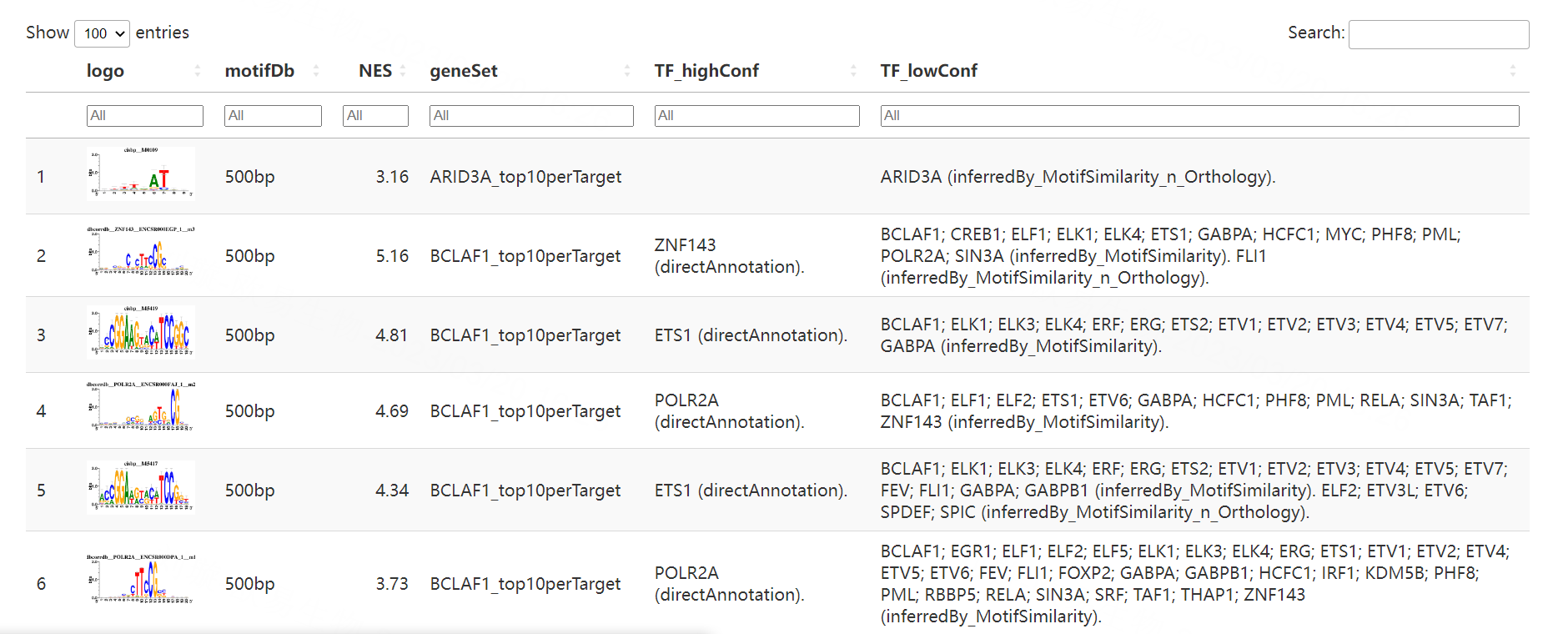
3. Regulon活性定量
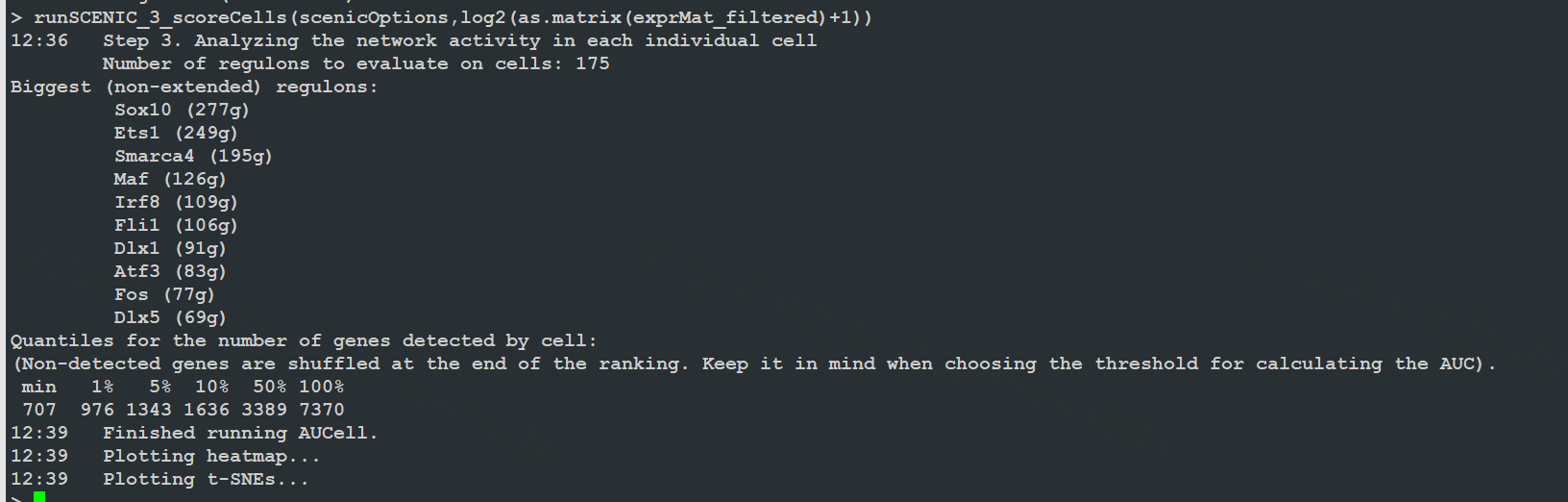
上一步我们已经获得regulon了,这一步我们针对以Regulons中所有基因作为基因集,针对每个细胞,将细胞中所有基因按照表达量从高到低进行排序,根据Regulons中的基因在排序中的位置,计算累计曲线面积 (AUC) ,即为Regulons在细胞中的活性。这步输出是以regulon为行,细胞为列的这样一个矩阵。其实这种矩阵对细胞类型清晰的数据集已经够用了。但对于细胞类型不清晰的数据集,SCENIC建议可以将AUC矩阵进行二值化,使用混合高斯模型,根据AUC值的双峰分布特征计算阈值,评估regulon处于on还是off的状态。二值化的AUC矩阵可以最大程度的体现细胞间的差异,可以用于细胞聚类,消除技术偏移,甚至消除跨物种比较的差异。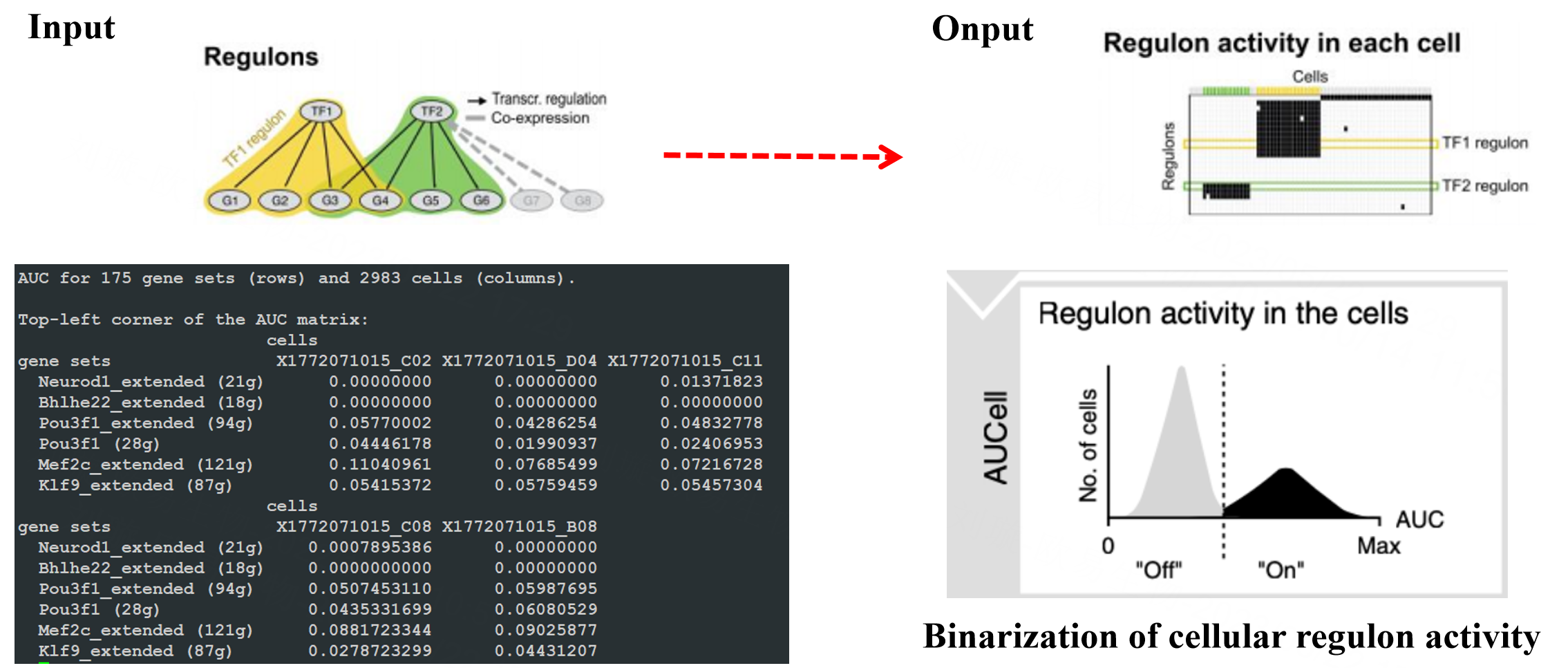
这一步我们首先需要重新运行intilalizeScenic,将并行处理取消,也就是将nCores设置为1。 然后运行runSCENIC 3 scorecells。运行之后将AUC矩阵保存回seurat对象的SCENIC slot中。可以看到这步可以生成以3开头的中间文件,而且SCENIC会使用AUC矩阵自动进行tsne的降维聚类,结果输出在output文件夹中的step3系列文件。
1 | |
1 | |
如果需要对AUC进行二值化,可以使用runSCENIC_4_aucell_binarize。使用该函数会在int文件夹下生成以4开头的rds文件及output文件夹下的Step4系列文件 。可以看到这次生成的文件就是二值化的AUC矩阵。 如果AUC值小于计算得来的阈值,则判定为该Regulons在该细胞中未开放,即未发挥调控作用。最终获得每个Regulons在每个细胞中的开放性热图。这里我们不进行展示了。
Tips: regulon有两种写法,第一种是TF名称+extended+靶基因数目,即转录因子与所有靶基因组成的基因调控网络。第二种是TF名称+靶基因数目:转录因子与高可信度靶基因组成的表达网络。
结果可视化
结合Seurat对SCENIC的结果进行图片展示
可视化的方式比较多,但使用到的都是生成的3.4 regulonAUC.rds的矩阵。
- 按照celltype分组绘制regulon平均活性的热图。
先把seurat对象的Idents改成meta.data里想要的分组变量列,我们这里是celltype。然后将分组信息以数据框dataframe的格式保存到CellInfo中。使用LoadInt加载AUC矩阵,使用onlyNonDuplicatedExtended对regulon进行过滤。然后我们对AUC矩阵分组计算平均AUC值,使用scale函数进行归一化,然后使用complexheatmap进行热图的绘制。
Tips:
onlyNonDuplicatedExtended的作用:如果同时存在高置信度的regulon和转录因子与所有靶基因组成的regulon,就仅保留高置信度的regulon。
降维图
可以使用Seurat的降维结果对感兴趣的regulon进行展示 ,展现一个映射AUC值的umap降维图。读入AUC矩阵,使用readRDS直接读入的矩阵是aucell包自带的文件格式,需要使用AUCell::getAUC进行转化。最后,将AUC矩阵加回seurat对象的metadata中,将我们感兴趣的调控因子映射到Umap降维图中。小提琴和山脊图。
流程和之前类似,都是读入AUC矩阵,使用AddMetaData函数将AUC矩阵加回到seurat的metadata中。分别使用Ridgeplot和vlnplot绘制山脊图和小提琴图。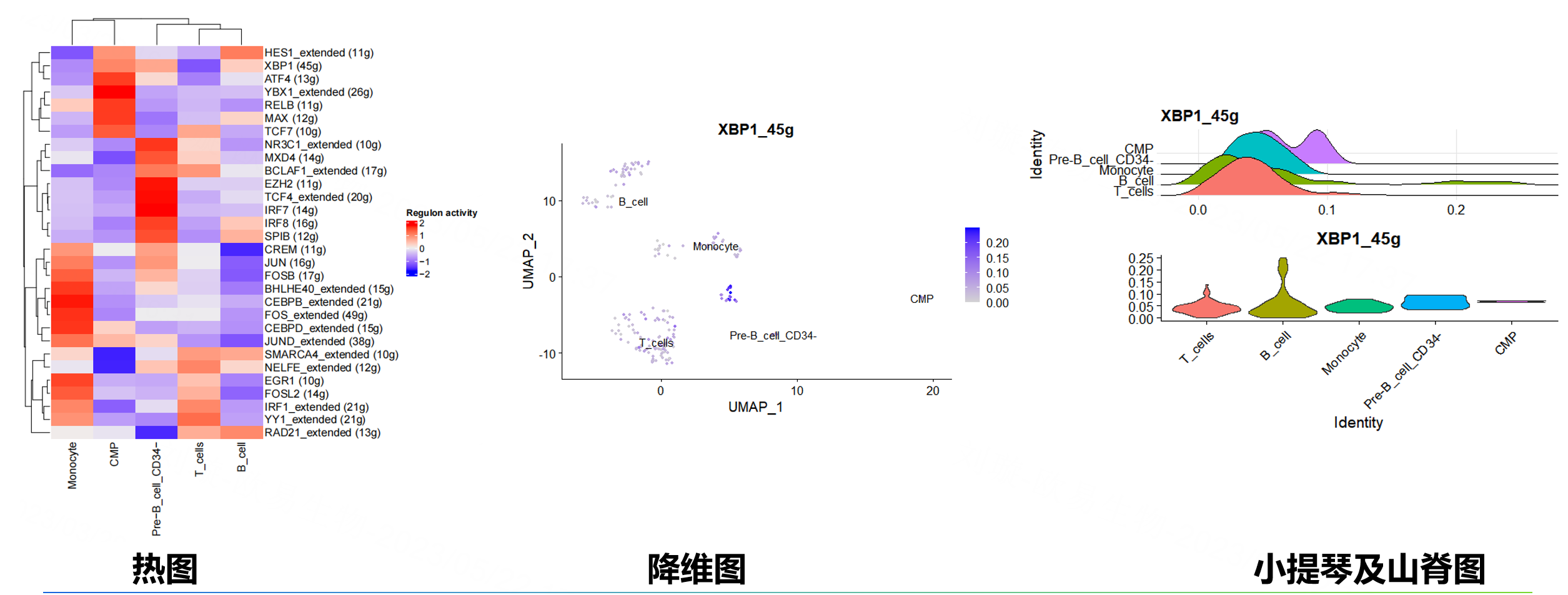
Regulon specific score(RSS)
Regulon specific score(RSS)预测了regulon与每种细胞类型的特定对应关系,RSS值越高表示该regulon在对应的细胞类型中特异性越⾼。
RSS需要两个vector进行计算,PR反应的是RAS在所有细胞中的分布。针对单个细胞,RAS就是选定regulon对应的AUC值,⽽针对每个细胞群,根据细胞的RAS,剔除细胞群中
活性得分超出中位值±3倍mad值的(异常值)细胞后,以平均值代表该细胞类型的RAS。对PR进行标准化。
然后用另一个vector PC来判断一个细胞是否属于一个特定的细胞类型,若属于则赋值为1,反之赋值为0。 这就是代表一种极端理想的情况,就是选择的regulon仅在这一个细胞类型中具有活性,也就是说是这种细胞类型独有的regulon。对pc同样进行标准化。
然后计算JSD值来评估两个vector之间的差异。JSD的值在0到1之间,0表示两个vector代表的分布完全相同,1表示两个分布完全不同。RSS的计算公式如下。RSS值越大,JSD值越小,计算的regulon在选定的细胞类型中特异性越高。
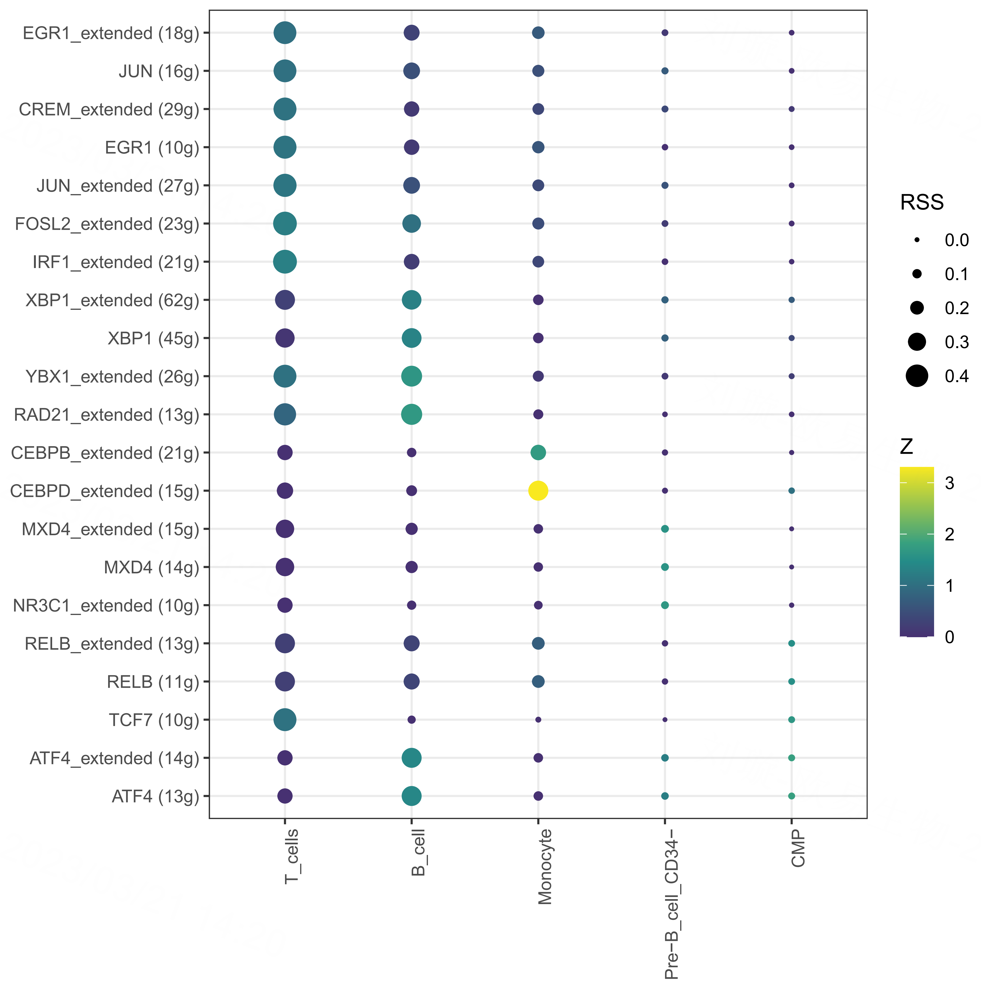
颜色深浅代表zscore值,点的大小代表rss评分。绘制只需根据seurat对象中的细胞类型信息,计算一下RSS值,然后使用PlotRSS函数就可以进行绘制了。由于我们采用的这个稳定版本的scenic没有内置aux_rss.R的函数(这个函数是封装在Pre-release,也就是内测版的scenic 1.3.0中),所以是使用source的方式进行R脚本的调用的。
下图为RSS_ranking_plot,横坐标表示排名,纵坐标表示RSS得分。RSS越高的调控子可能与该细胞群特异性相关。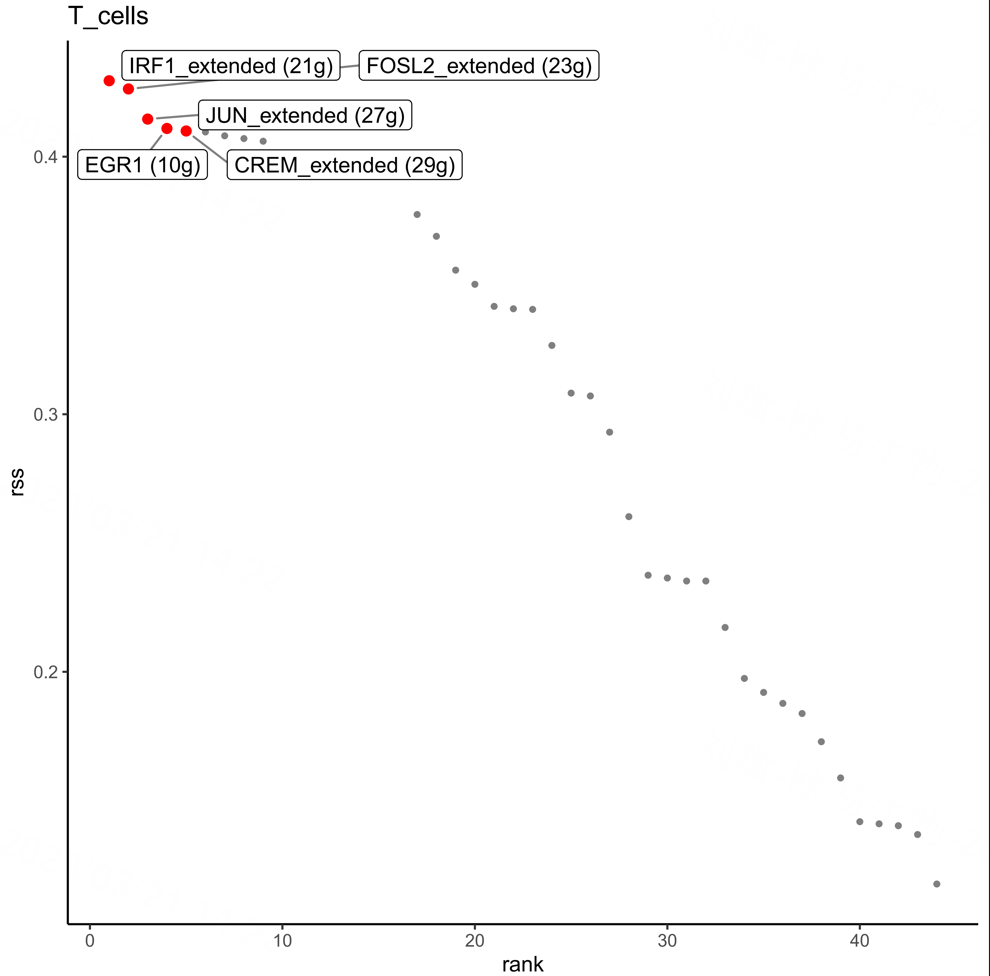
针对感兴趣的细胞类型查看细胞特异性Regulon。前面的绘图准备工作都和plotRSS是一致的,只不过这一步使用到的函数是plotRSS_onSet(可以从scenic的github查看,我们这里是直接把函数拆分了进行画图),输入rds,设置想要看到多少个细胞特异性regulon,我们这里是设置成了五个,然后使用setName设置感兴趣的细胞类型,绘图即可。
Connection Specificity Index(CSI)
第二个在scenic分析中常用的指标是CSI, 该指标用来划分regulon module,展示不同Regulon之间的关联性,同时具有较高CSI的Regulon可能共同调控下游基因,并共同负责细胞功能。
CSI 的评估包括两个步骤,⾸先计算regulon配对的pearson correlation coefficient(PCC),接下来对于选定的⼀对regulon pair,假设A和B,得到A与B的PCC值,针对其他与A或B相关的配对,统计其中PCC值低于A与B之间PCC值的⽐例,这⼀⽐例就是A与B配对的CSI值,CSI值越⼤表示regulon 配对中的两个regulon相关性越⾼。最后,对所有regulon配对的CSI矩阵进⾏层次聚类(Hierarchical clustering),划分regulon module,对于每⼀个regulon module,其在每种细胞类型中的活性得分等于该细胞类型内所有细胞类型对于该module内所有regulon的RAS的平均值。
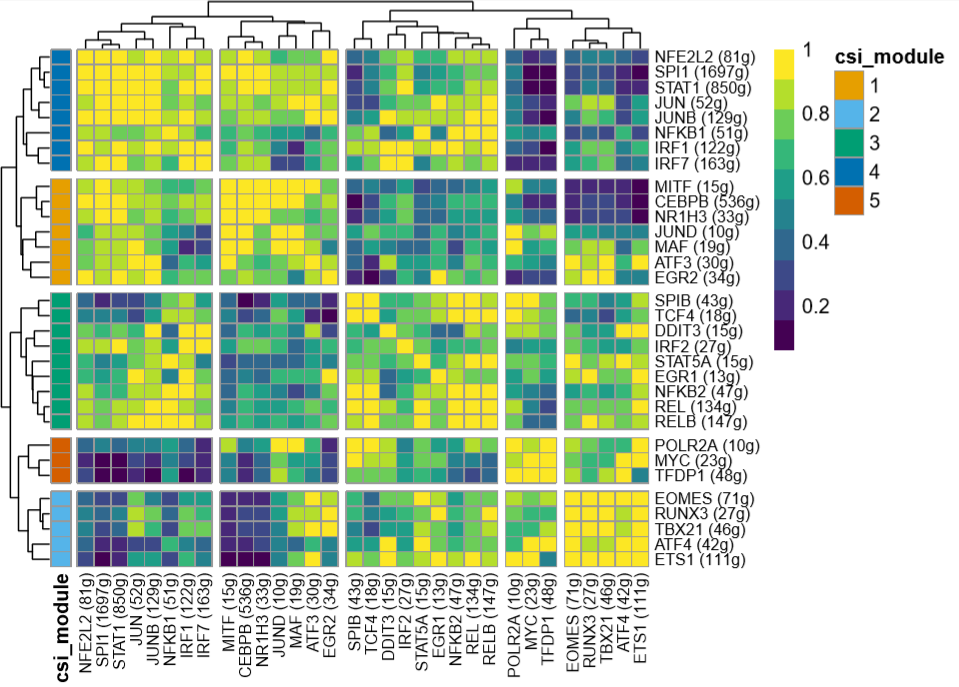
各细胞群中CSI关联模块的活性热图。行表示根据CSI聚类热图进行手动划分的CSI模块,列表示不同细胞群。颜色由蓝变黄表示CSI模块活性由低到高。活性相似的CSI模块对应的细胞群可能具有相似的基因表达模式以及相似的调控网络。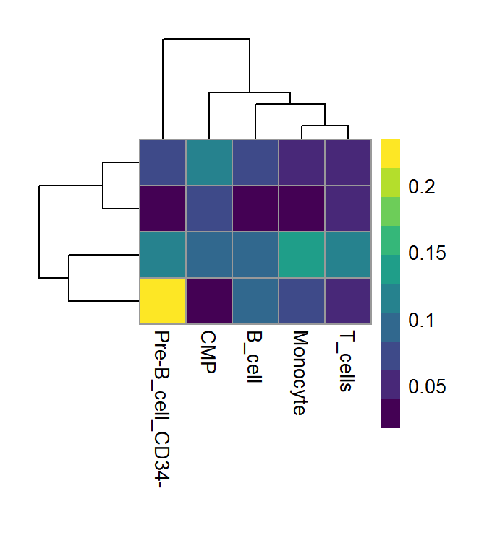
美化后的CSI关联模块的活性热图如下(请结合美化后的Regulon模块的CSI关联性聚类热图共同查看):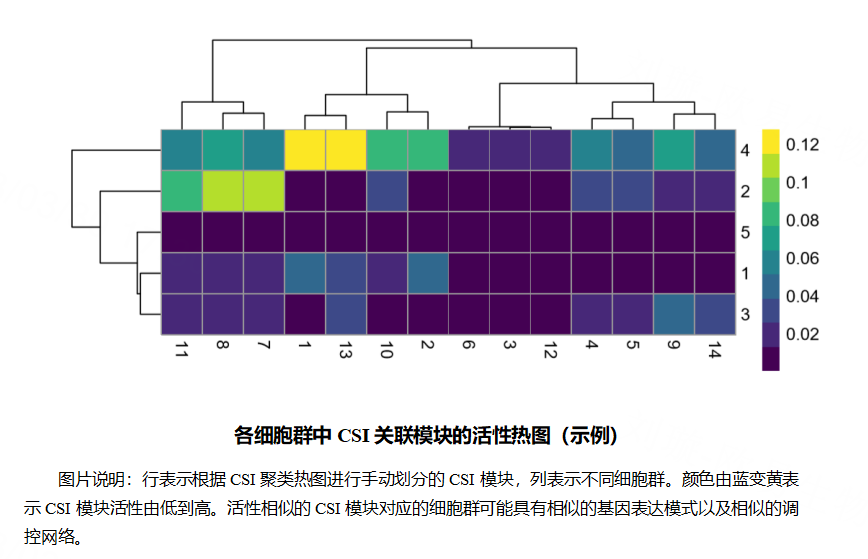
下图为Regulon模块的CSI关联性聚类热图,行列均表示regulon,颜色由蓝变黄表示CSI关联性值由低到高。CSI值都较高的regulon可能具有相似的细胞功能,共同调控下游基因。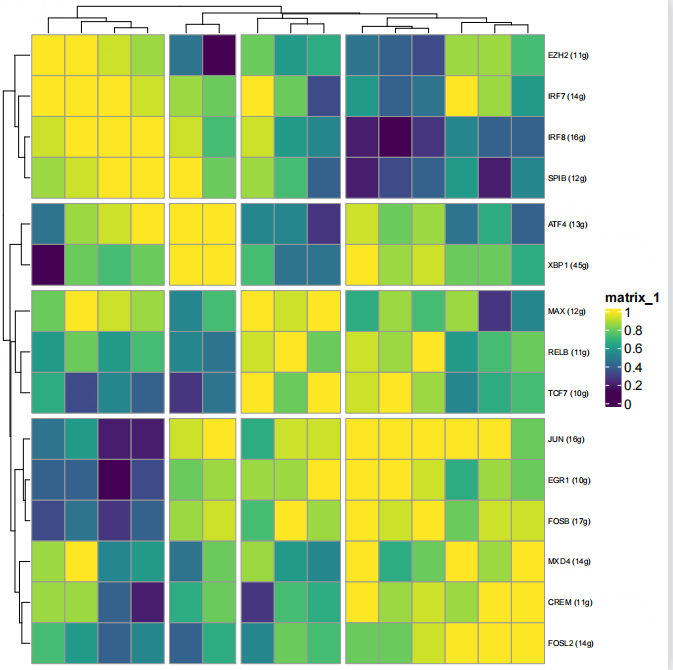
美化后的Regulon模块的CSI关联性聚类热图如下图,