Cellchat
Cellchat的学习笔记及代码分享
细胞通讯背景介绍
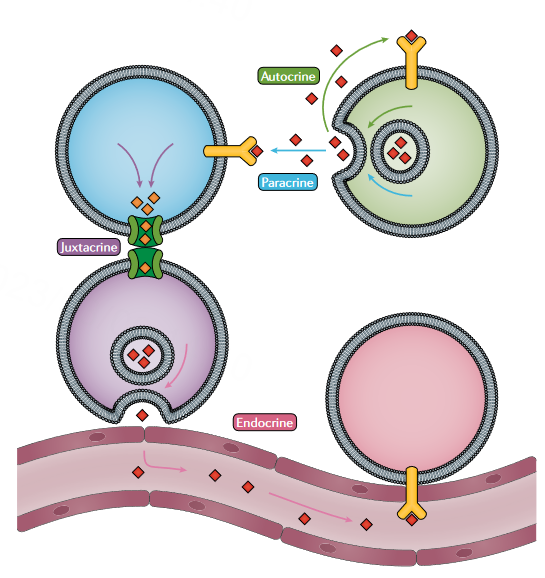
细胞通讯主要由一个信号产生的细胞发出的信息,一般是通过一个介质,也就是配体,传递给靶细胞(如图Y就是受体)。配体和受体之间是特异性结合,靶细胞接触配体后就会产生一系列的生理生化效应。
自分泌(Autocrine)
是指同一个细胞内的通讯,细胞通过分泌配体来诱导自身细胞反应。旁分泌(Paracrine)
不需要细胞-细胞接触,而是依赖于分泌后信号分子从一个细胞扩散到另一个细胞。近分泌(Juxtacrine)
细胞间通讯依靠缝隙连接或其他结构如膜纳米管直接在细胞间传递信号分子,需要细胞相互接触。信号分子不分泌到细胞外空间。内分泌(Endocrine)
信号分子通过细胞外液(如血浆)被分泌并长距离传播;这种交流的典型介质是激素。
细胞通讯参与许多生物进程,理解细胞通讯可以帮助我们了解细胞与细胞之间的互作关系。通过选择不同时间点的样本或同类细胞的不同亚群可以揭示发育过程中各类细胞的相互作用。细胞通讯可以用于探索肿瘤免疫微环境,比如先从细胞亚群入手,通过对不同亚群进行受体-配体分析,挑选相互作用最强的亚群,然后探索该亚群与其他细胞类型的相互作用,揭示细胞亚群在肿瘤免疫微环境中的作用机制。也可以用于挖掘疾病治疗靶点等。
Cellchat
Cellchat简介
单细胞领域,2021年美国加州大学开发出了cellchat软件用于分析细胞通讯分析。cellchat这个工具具有非常强大的灵活性,不仅可以对细胞通讯网络进行推断和分析,还具有丰富的可视化功能。
- 使用单细胞表达谱与已知的细胞通讯数据库进行细胞通讯互作强度的计算。
- 通过配体-受体的互作概率与扰动检验,识别显著互作的配体-受体关系对。
- 通过加和细胞类型间显著互作的配体-受体关系对数量或者强度来计算和整合细胞间通讯网络。
虽然同样是从scRNA-seq数据中推断细胞间通讯,但Cellchat与NicheNet等只使用一个配体/受体基因对的软件不同,Cellchat将配体-受体+多聚体+辅助因子同时发挥作用的情况纳入考量。这就考虑到了比如细胞因子TGF β在TGFBR2/TGFBR1二聚体的参与下调节细胞增殖这种常见的情况。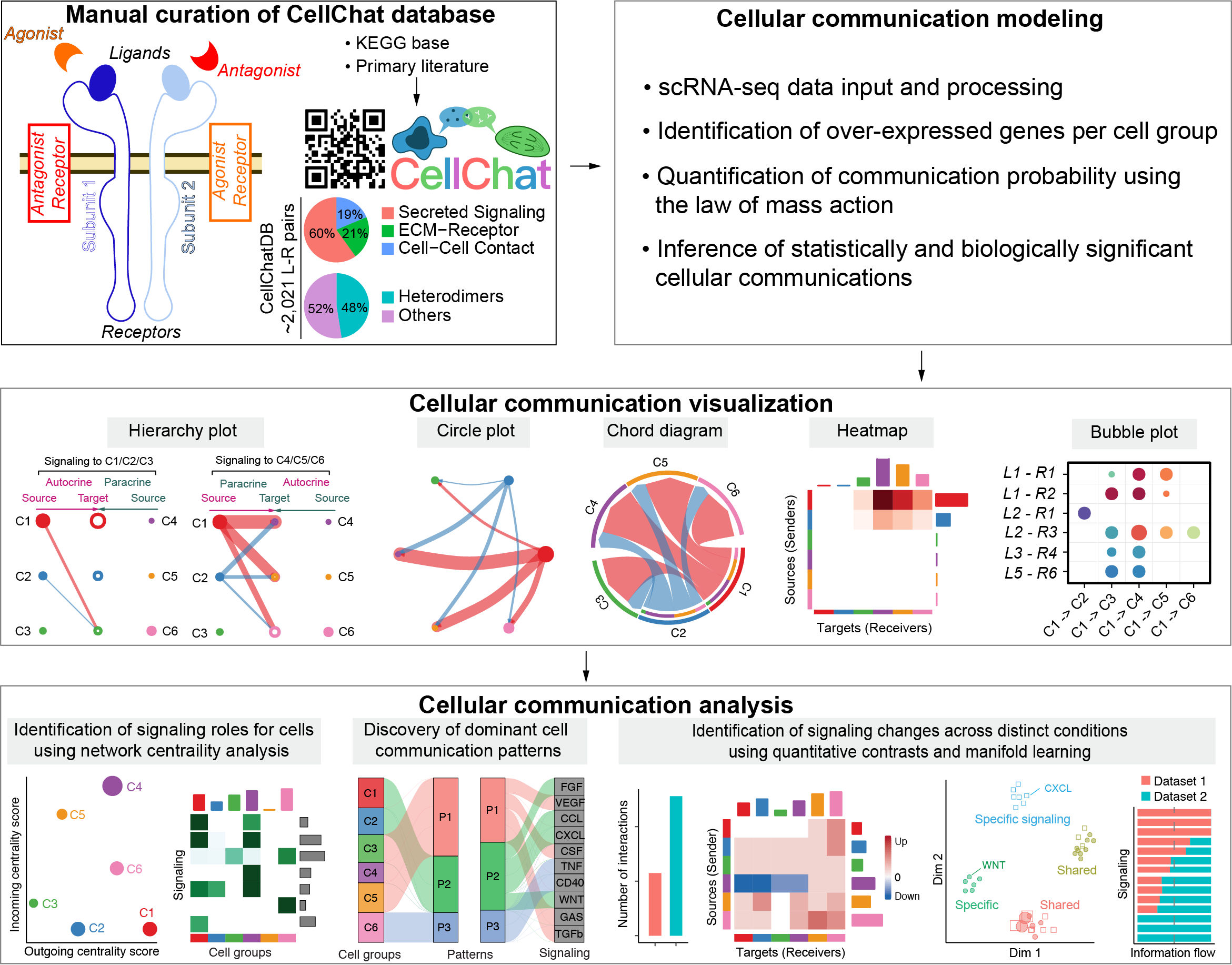
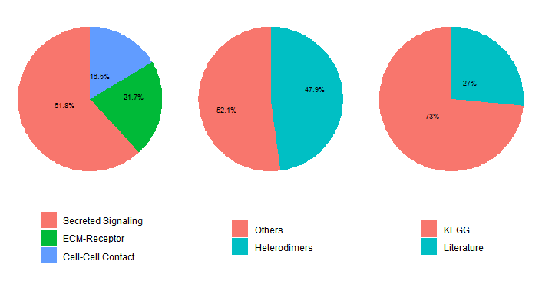
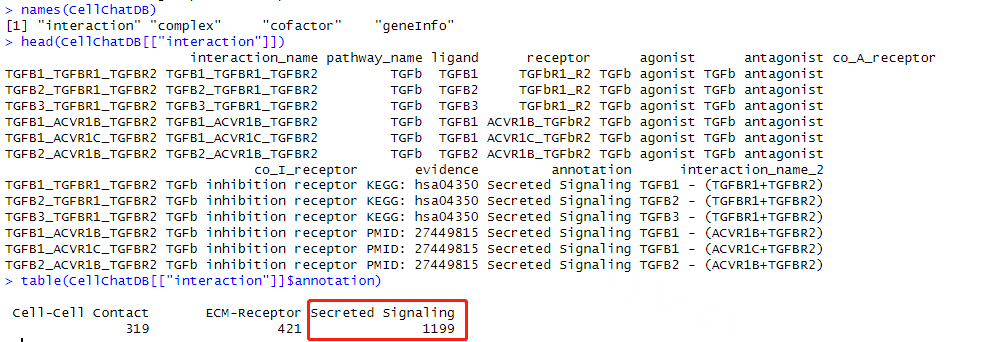
Cellchat强大之处在于它的数据库, CellChat 软件构建了一个配体、受体及其辅因子间相互作用的数据库——CellChatDB,cellchat数据库是人工注释的数据库,支持人和小鼠受配体互作推断,在其github里发现开发团队更新了斑马鱼的数据库,用户也可以根据自己获得或自己挑选的数据,用cellchat自带的功能对它的数据库更新并使用。这个代码可以帮助用户看的它的数据库组成,我们在后面进行分析时,可以根据需求筛选特定的数据库进行分析。根据通讯方式不同,可以将信号通路分为以下三种:Secreted signaling主要指的就是内分泌,自分泌和旁分泌;Cell-cell Contact主要对应近分泌;ECM受体主要是整合素等其他细胞表面相关成分,通过细胞与细胞外基质接触来介导细胞通讯(是一种特殊的近分泌)。
提供了Web端可用于搜索受配体
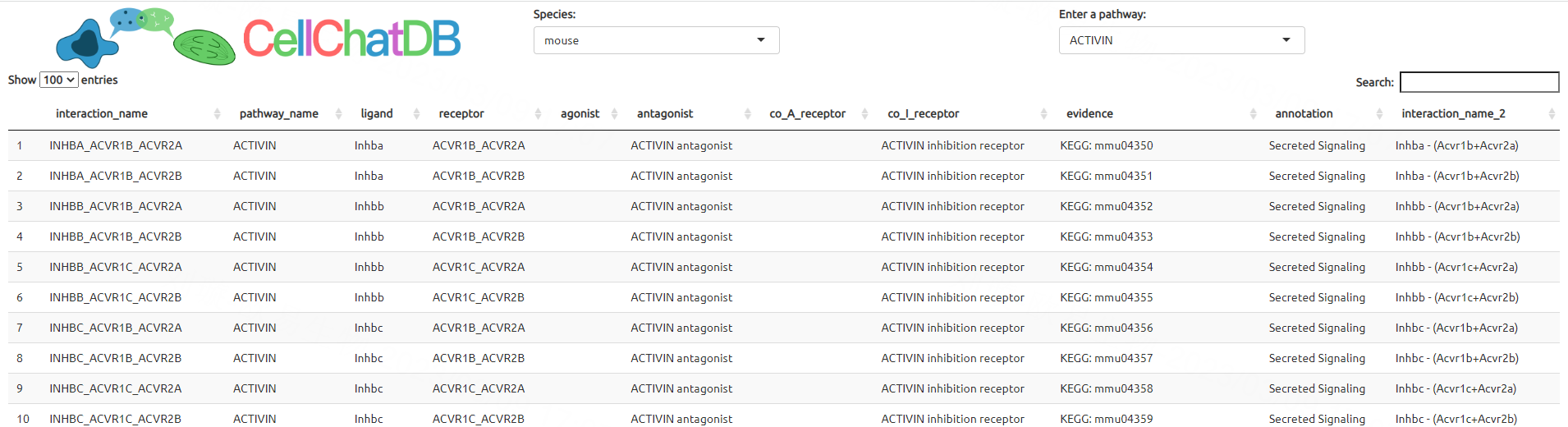
#以human为例查看数据库构成
showDatabaseCategory(CellChatDB.human)
Cellchat 分析原理
我们要看细胞亚群之间的通讯作用,首先仍然是需要进行差异分析,计算哪些基因是在细胞群之间存在差异表达,这是通过identifyOverExpressedGenes函数实现的。cellchat使用Wilcoxon检验,在显著性水平为0.05的情况下,在所有细胞组中识别差异表达的信号基因。然后这些过表达基因所在的互作即过表达互作。通过identifyOverExpressedInteractions函数可以识别这些过表达互作,并将他们存于cellchat@LR$LRsig。
第二步是对不同的细胞群计算一个平均表达水平(使用了基因表达分位数的方式,排除了异常值的影响,公式见文献)
第三步也是最重要的一步,就是对每个配体受体对计算通信概率。
配受体对表达情况计算:
Li代表配体L在细胞群i中的表达水平。如果配体L有多个亚基,计算的是几何平均数作为整体的表达水平:也就是任何一个亚基的零表达都会导致一个无活性的配体。
Rj代表受体R在细胞群j中的表达水平。如果有多个亚基,仍然计算几何平均数。比较特殊的是,由于共刺激和共抑制膜结合受体能够通过控制受体激活来调节信号,因此在计算受体时,会将共刺激受体纳入考量。
对于具有多个共刺激受体的配体-受体对,我们计算了这些共刺激受体的平均表达量(用RA表示),然后使用线性函数来建模对受体表达的正向调节(activating)。(1+RA)
对于每个具有多个共抑制受体(RI)的配体-受体对,我们使用相同的线性函数方法对它们进行建模。(1+RI)
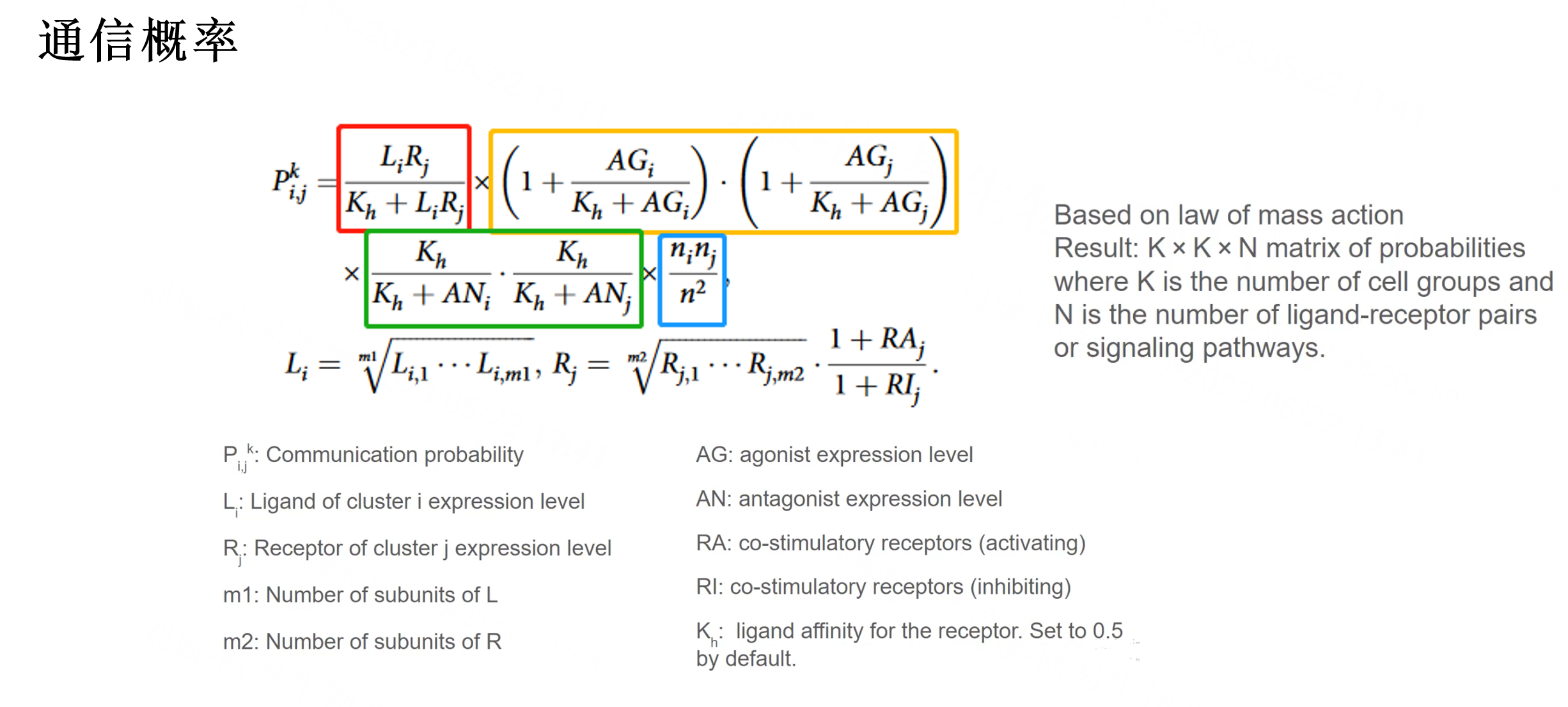
通讯概率计算:
红色框中的部分即为使用Hill系数对受配体之间的相互作用进行建模,参数Kh的默认值设为0.5,因为输入数据的归一化范围从0到1。(希尔系数主要是用于研究蛋白质内部,如果是由多亚基组成,那么这一群亚基之间有怎样的作用关系,这里是取它的延伸作用。 Kh在希尔方程中常作为解离常数,来源于质量作用定律)
来自发送细胞和接收细胞的胞外激动剂和拮抗剂能够直接或间接地调节配体-受体相互作用:
黄色框展示了对激动剂的建模。
对于含有多个激动剂的配体-受体对,我们计算了这些激动剂(agonist)的平均表达量(用AG表示),然后使用Hill函数来模拟激动剂对配体-受体相互作用的正调节。
原理相似,绿色框中的部分为对拮抗剂的建模(antagonist)。
蓝色框这部分会将每个细胞群中细胞比例的影响也被纳入概率计算中,其中ni和nj分别为细胞群i和j中的细胞数量,n为给定数据集中的细胞总数。
这样我们就可以计算出来所有配体-受体对之间的所有细胞组之间的通信概率, 这个整体的通讯概率由一个三维array ( K × K × N)表示,其中K是cell group的数量,N是配体-受体对或信号通路的数量。
我们比较关心在统计学上,这些通讯是否具有显著性呢?
cellchat会基于置换检验的方法,将细胞标签打乱重排一百次, 生成零分布,计算p值。
最后通过层次图(hierarchy plot),圈图(Circle plot),气泡图(bubble plot)等多种方式进行结果展示。
CellphoneDB
CellphoneDB 简介
CellPhoneDB 构建了一个公开的物种为人类的经过人工注释的受体、配体及其相互作用的数据,并配有相应的R包和python包来对单细胞转录组学数据进行分析。
与cellchat相似,cellphoneDB考虑到了异聚复合物。在存在多个亚基时,表达量是按照最低的亚基进行计算,也就是说cellchat和cellphoneDB都会因为多聚体的任何一个亚基表达量为0而导致多聚体的表达量按0进行计算。cellphoneDB的相互作用注释也比较全面,有两千多种相互作用,同时也支持自定义受配体相互作用。
与CellPhoneDB v3相比,CellPhoneDB v4.1版本的可信度更强,因为它不再从外部资源导入交互,而是增加了更多的人工注释条目,并考虑了非蛋白分子作为配体的情况。由于开始支持python,运行速度也得到了大幅提升。
CellphoneDB原理
CellphoneDB要求输入normalized后的矩阵(data矩阵),在给定表达矩阵和细胞注释之后,对于细胞群中的每个基因,在数据集中的所有原细胞分群之间执行成对比较,计算表达该基因的细胞百分比和基因表达的实际平均值。
然后,随机排列所有细胞的细胞类型标记,形成新的细胞群(默认随机排列1000次),计算随机排列后细胞群中配体的平均表达水平和与其相互作用的细胞类型中受体的平均表达水平的平均值。通过这种方式,在两种细胞类型之间的每对配对比较中为每个配体-受体对生成一个零分布(null distribution)。
生成零分布之后,根据计算平均值等于或高于实际平均值的比例推测该受体-配体对在这两种细胞类型中可能的显著性P值。
之后,根据两种细胞类型中富集到的显著的配体-受体对的数量,对细胞群中显著的受配体进行排序,找到特异性的配受体,以便手动筛选出生物学相关的相互作用关系。这样,通过特异性的蛋白复合物,预测了细胞群之间可能的分子相互作用,产生了细胞群间潜在的通讯网络,这些网络可以通过直观的表格和图表进行可视化。
CellphoneDB的下游可视化相比cellchat要少,可以将生成的表格导入Cytoscape进行网络图的可视化。
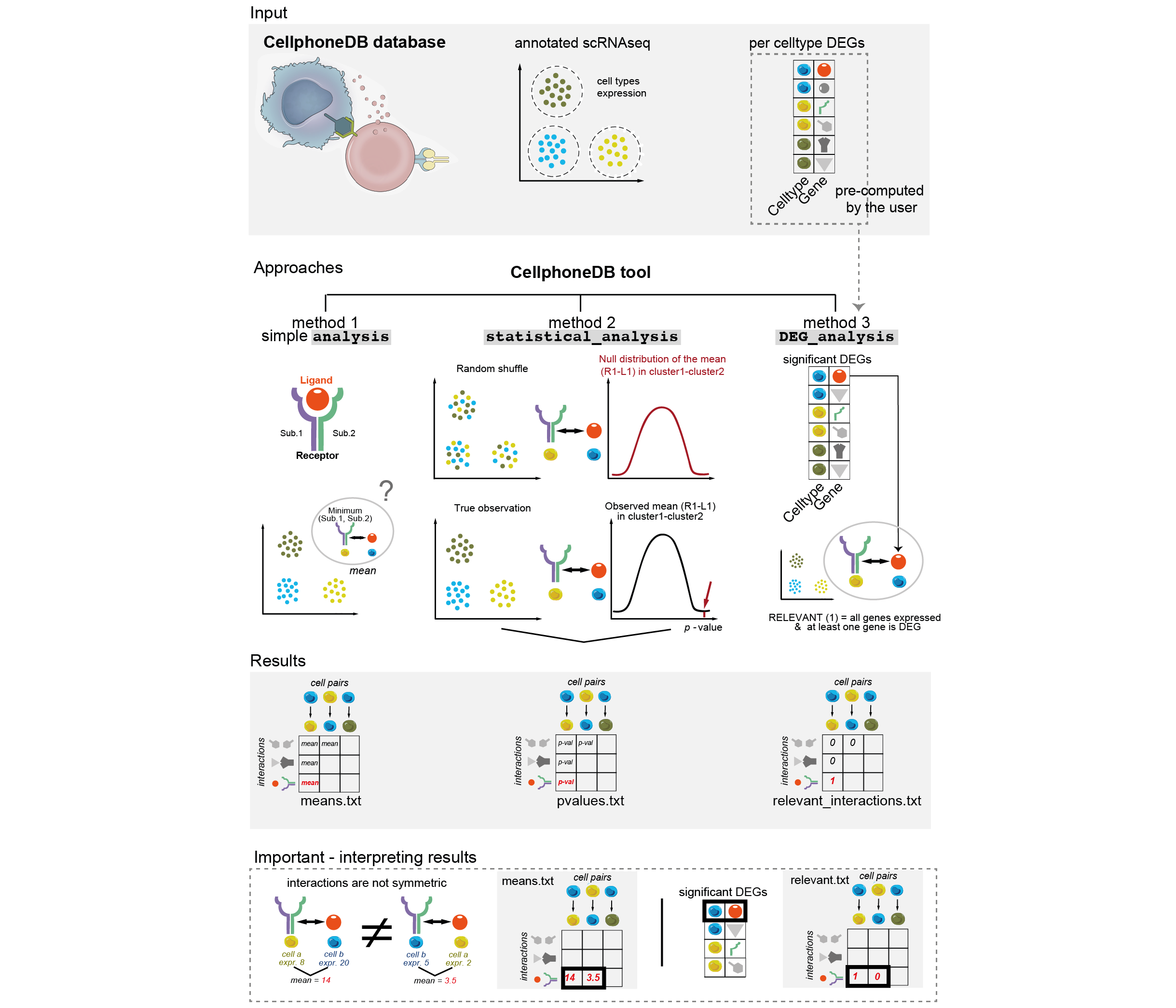
CellphoneDB与Cellchat比较
CellphoneDB:
- 基于 人 开发,可通过同源转换应用于小鼠
- 考虑二聚体
- CellphoneDB数据库包含将近3000种相互作用。
- 考虑200多种涉及带注释的非肽分子(即不由基因编码),例如类固醇激素,的相互作用。
- CellphoneDB v3支持空间信息
- 只有表达受体和配体基因的细胞占比超过指定的阈值时(默认为10%),该配体-受体对才会被纳入分析。
- 基于Shuffle,运行速度相对较慢(v4.1基于python, 运行速度提升)
Cellchat:
- 基于 人 和 小鼠 开发
- 考虑二聚体和拮抗剂等
- 数据库中含有2021种已验证的相互作用,其中60%为分泌信号,48%的相互作用需要二聚体复合体参与。
- Cellchat v1.6.0支持空间信息
- 基于R开发
- 可以通过结合网络分析、模式识别和流形学习方法,使用集成方法定量表征和比较推断的细胞间通信网络。
Cellchat 代码详解
数据准备
首先要准备分析数据,也就是构建cellchat对象。我们需要表达矩阵和对应的标识信息两种数据,我们用常见的seurat对象为例,这里用的是我们自己的示例数据,只用于展示分析步骤和产生的结果。
在读取seurat对象之后,建议将我们需要用于推断互作关系的细胞类群对应的标签设置为seurat对象的ident方便后续转换。cellchat提供了不同的转换方式:
方案一:我们可以从seurat对象中提取表达矩阵以及细胞对应的meta信息。之后通过createCellchat这个函数手动构建cellchat对象,运行了createCellchat函数之后终端会提示我们从数据矩阵构建的cellchat对象,并且提示我们采用的是哪些细胞类型进行细胞通讯分析。
方案二:createCellchat这个函数也支持直接将seurat对象转换为cellchat对象。运行之后也可以看到终端提示我们构建数据的来源和即将进行分析的细胞类型。同时我们可以看到,我们之前给seurat对象设置的ident也被添加到cellchat对象的meta信息里了。
1 | |
设置数据库
进行细胞通讯分析需要先将数据库添加到cellchat对象中,根据我们样本物种选择对应的数据库,可以使用全部的数据进行计算,也可以根据需要筛选部分数据,将数据添加到cellchat对象的DB slot里。由于是demo数据演示,故只选取了动植物细胞中最常见的通讯方式,也就是分泌信号的数据库,在实操的时候可以改为完整的数据库,看看还有哪些信号通路被纳入分析。
1 | |
数据预处理
官方提示不管是不是需要提取cellchat子集都需要跑subsetdata这一步,其实这一步也是在对我们的数据本身的基因与数据库中的基因取交集并且将对应基因的表达矩阵保存到名为data.signaling 的slot里面,所以需要跑这一步来节约计算成本是必要的。在识别过表达基因(也就是差异分析)和过表达的受配体对之后,(鉴定与每个细胞组相关的过表达信号传导基因(var.features slot)、识别所用cellChatDB中过度表达的配体受体相互作用(对)(LR slot))。
1 | |
细胞通讯概率计算
首先使用computeCommunProb函数计算任何相互作用的细胞群之间的通信概率/强度,保存在net slot里面,里面包括细胞类型对和受体配体对相互作用的信息。此步骤运行稍微耗时。
在数据量较大的情况下,细胞两两配对的信息非常多,这里就需要一个筛选的过程,针对某个细胞群内细胞数目小于一定阈值,我们将与它相关的通讯过滤掉。这里使用filterCommunication函数并将阈值设定为10个细胞。
computeCommunProbPathway函数通过汇总所有相关配体/受体来计算信号通路水平的通信概率,并保存在netP slot中。
最后使用aggregateNet函数通过计算通讯数量或汇总通信概率(强度)来计算聚合网络。还可以通过设置源来计算单元组子集之间的聚合网络。使用souce.use和targets.use。
所有配体-受体对之间的所有细胞组之间的通信概率由一个三维阵列P ( K × K × N)表示,其中K是cell group的数量,N是配体-受体对或信号通路的数量,这个array就是cellchat@net$prob。net由配体/受体级别的所有推断细胞通信组成,而netP可以在信号通路级别访问推断的通信
1 | |
Cellchat可视化
总体展示
cellchat自带多种可视化的函数,整体来看各细胞类型直接的互作数量和强度,可以用网络图表示。
- Circle plot
1
2
3
4
5
6
7
8#使用circle plot显示任意两个细胞group之间的相互作用次数或总交互强度(比重)
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = groupSize, weight.scale = T,
label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize, weight.scale = T,
label.edge= F, title.name = "Interaction weights/strength")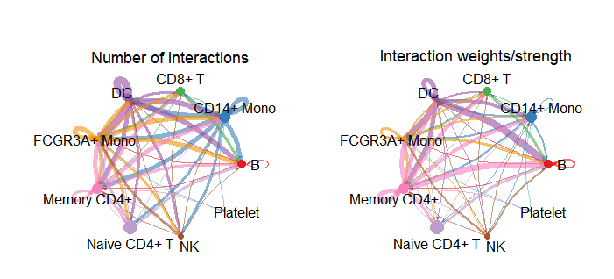
par()函数,是R基础绘图系统中一个比较常见的函数,在CellChat中主要是为了排列多个图形,例如这里的mfrow参数的形式就为c(nr, nc),表示将后续绘制的图形按照nr行nc列排布,而mfrow表示先横向后纵向,对应的也有mfcol表示先纵向后横向。xpd = True是设置允许在作图区域外作图,防止文字显示不全。使用netVisual_circle函数绘制的图展现在右侧,上图左侧展示了通讯的数量,右侧展示了通讯的强度,也就是概率值相加的结果。
可以看到我们设置了groupSize这个对象,反映了idents,也就是我们的分组变量,celltype的数量。因此,图上的圆圈大小就反应每个细胞类型里的细胞数的多少,线条的粗细反应了通讯的数量或者强度。
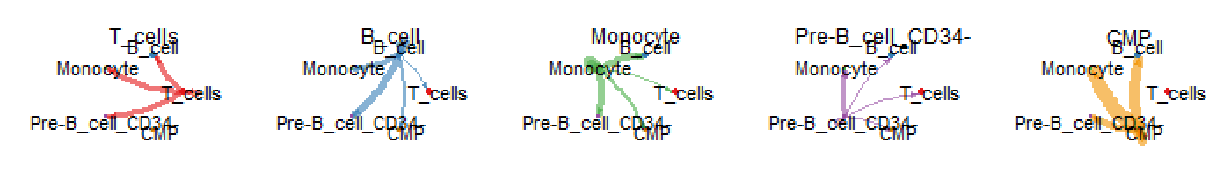
Tips: 单独展示
分别展示每个细胞类型与其他细胞类群的互作,采用的方式是将不包含该细胞类型的互作数量或者强度设置为0,在图片上就不会显示出来了。可以看”单独展示”这部分的代码。
1 | |
信号通路层面展示
信号通路层面的展示,也就是对netp数据槽的展示。
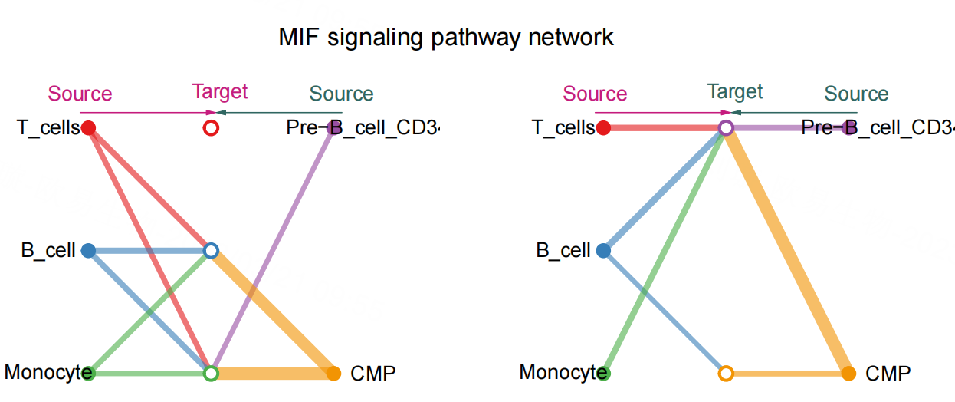
- Hierarchy plot 层次图
使用层次图可视化通信网络,请定义vertex.receiver,它是一个数字向量,给出作为第一个层次结构图中的目标的细胞组的索引。我们可以使用netVisual_aggregate来可视化信号路径的推断通信网络。
1 | |
在层次图中,实体圆和空心圆分别表示源和目标。圆的大小与每个细胞组的细胞数成比例。边缘颜色与信源一致。线越粗,信号越强。这里我们展示了一个MIF信号网络的例子。所有重要的信号通路名称都可以通过cellchat@netP$pathways访问。
Circle plot 环状图
1
2par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")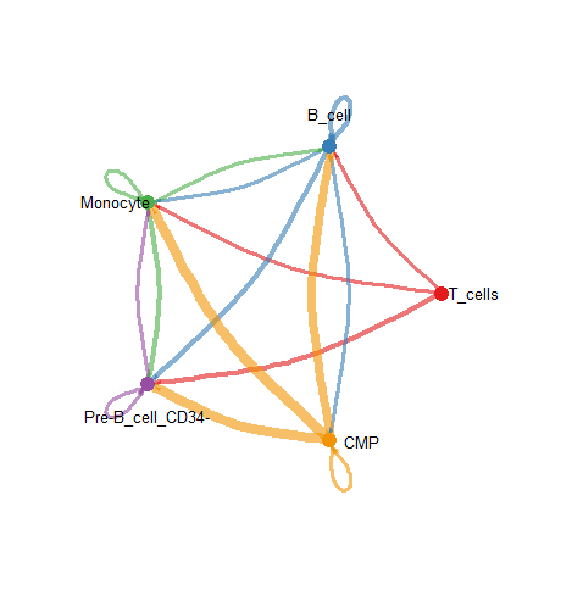
Chord diagram 和弦图
1
2par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "chord")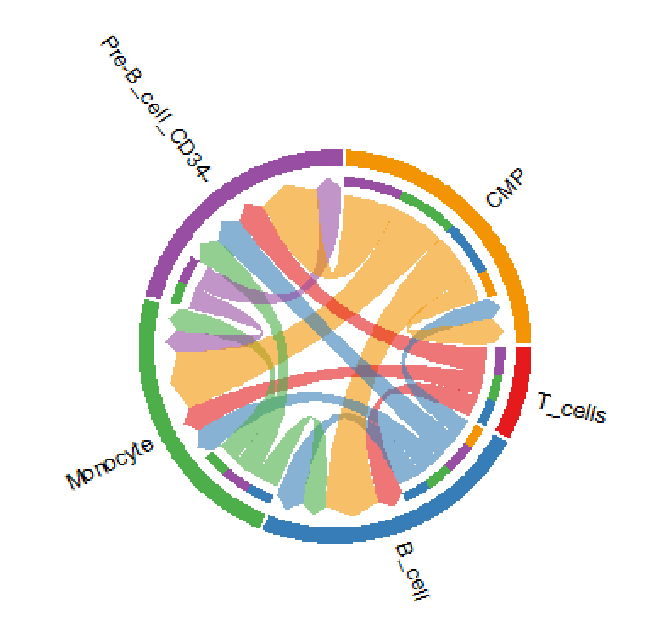
Heatmap 热图
1
2par(mfrow=c(1,1))
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")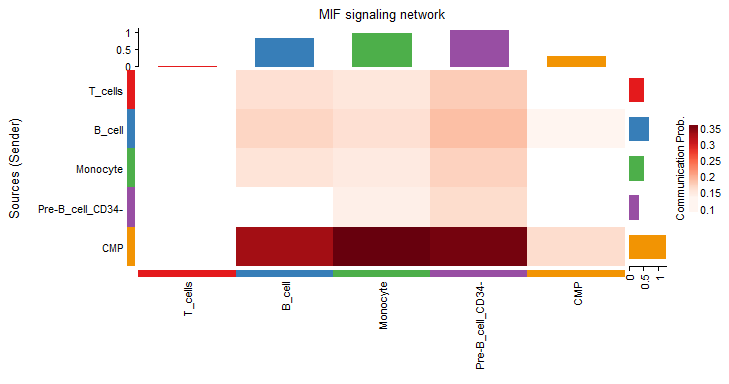
图左侧已经标明了信号发出的细胞群,上方和右方的柱状图分别表示每一列和每一行数值的总和。
社会网络分析
- 计算网络中心性评分
对于细胞通讯网络进行系统分析,便于我们系统的解释这些复杂的网络。
使用网络分析中的中心性度量来预测特定细胞类型的关键传入和传出信号,以及不同细胞类型之间的协调响应,可以方便地识别细胞间通信网络中的主要发送者sender、接收者receiver、中介者mediator和影响者influencer。基于计算的MIF信号网络的四个网络中心性指标,Heatmap显示了每个细胞群的相对重要性。
首先对netp计算网络中心性评分。
1 | |
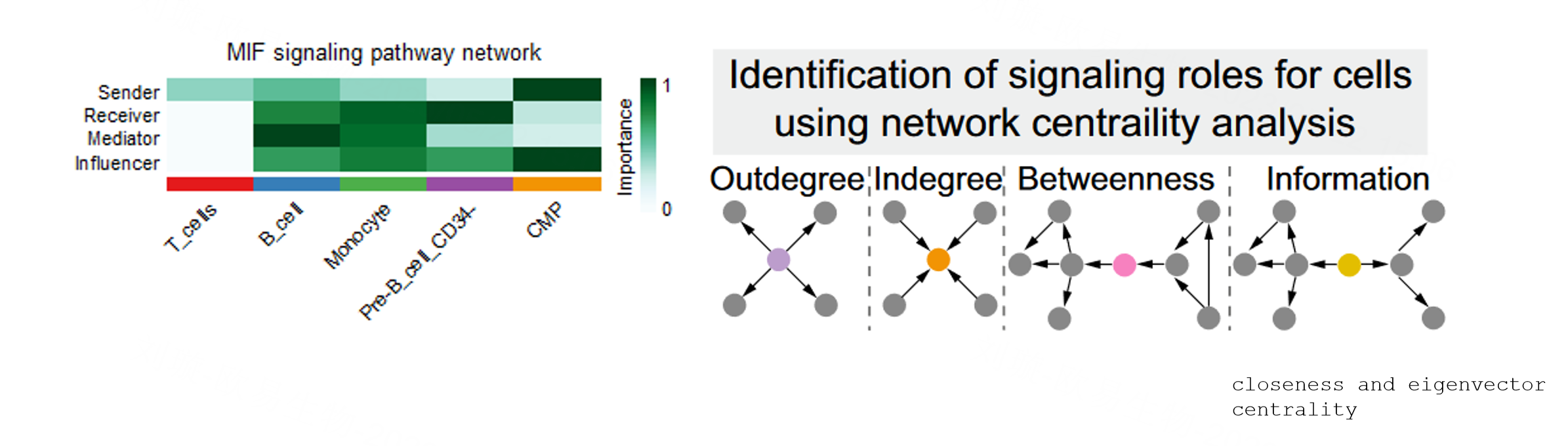
这块涉及到了网络分析的一些概念。Cellchat使用加权有向网络的测量方法,包括出度outdegree、入度indegree,中介中心性Betweenness和信息中心性Information,上图右侧的模式图是对这些概念的直观展现。
outdegree是对出度的衡量,对应着sender,计算的是发出信号的通信概率之和,直观展现就是从紫色圆圈发送信号到周围。
indegree是对入度的衡量,对应的是receiver, 橙色的圈接受从四周灰色的圈发出的信号。
betweenness centrality来衡量中介中心性,对应着mediator,可以直观的看到粉色圈圈承担了桥梁的作用,就是这个节点相当远一个闸,和它相连的节点想要到其他节点都得经过它。
Information这部分也是对中心性的一种度量方式,它使用接近中心性 和 特征向量中心性 的混合度量,综合考虑邻居节点多少和与旁边的节点有多接近,值越高反应对信息流的控制越强。
- 热图可视化
可以使用热图对特定的信号通路的入度和出度进行可视化,识别对某些细胞类群的输出和输入信号贡献最大的信号。
1 | |
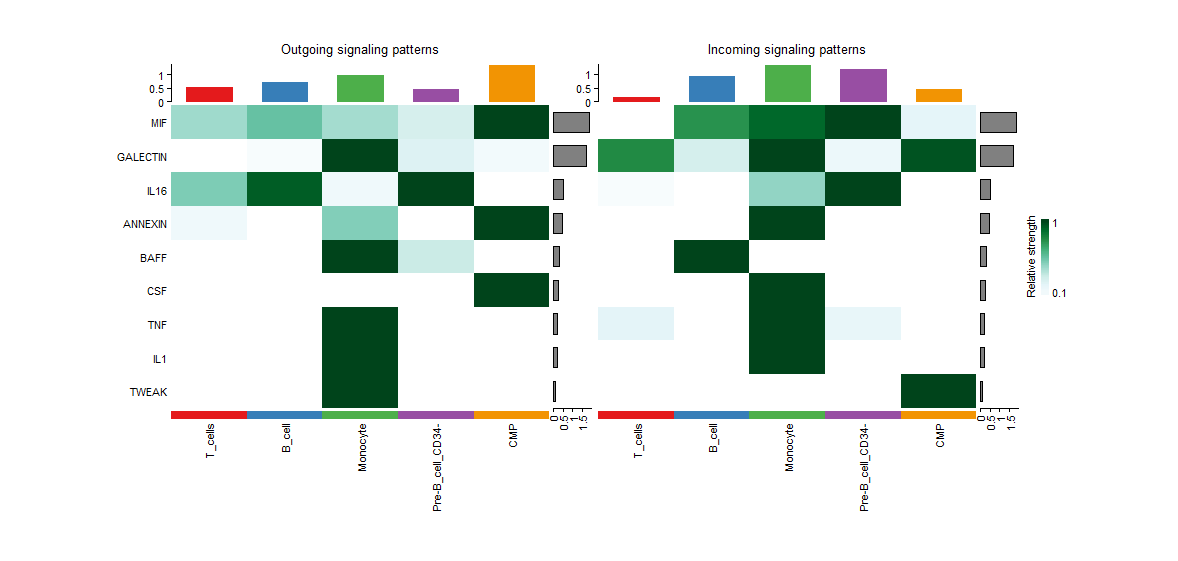
通讯模式分析
除了探索单个通路的详细通讯外,一个重要的问题是多个细胞群和信号通路如何协调运作。
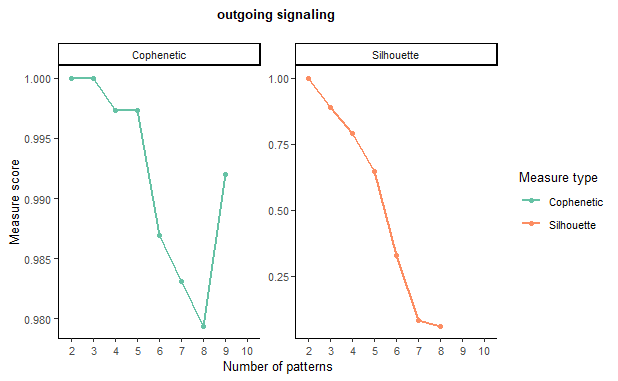
Cellchat调用了nmf包用于对信号通路和细胞群划分模式,功能相似的通路或者信号通路会被归类为同一种模式,通过pattern连接相应的细胞群和信号通路。随着模式数量的增加,可能会出现冗余模式,这使得解释通信模式变得困难。在出度的模式下,我们使用函数selectk来推断模式的数量,包括Cophenetic(共表型系数)和Silhouette(轮廓系数)。这两个度量标准都是基于共识矩阵的层次聚类来衡量特定数量模式的稳定性。
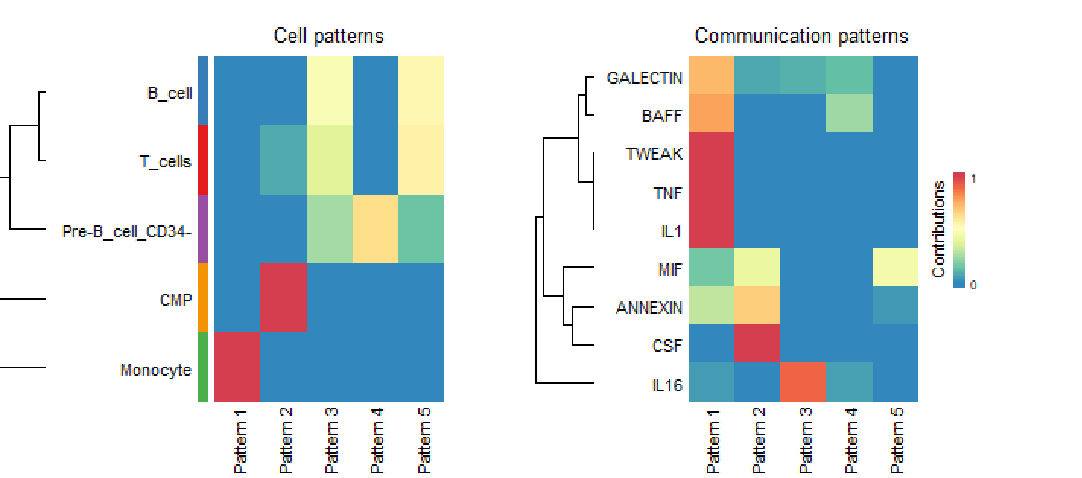
对于一系列模式的数量,一个合适的模式数量是Cophenetic和Silhouette值开始突然下降的前一个。这里我们来识别一下,这两个系数都是在6的时候明显下降,所以nPatterns应该设置为5,运行这个识别通信模式的函数,我们可以看到,类群和信号都被分为了五部分。
1 | |
1 | |
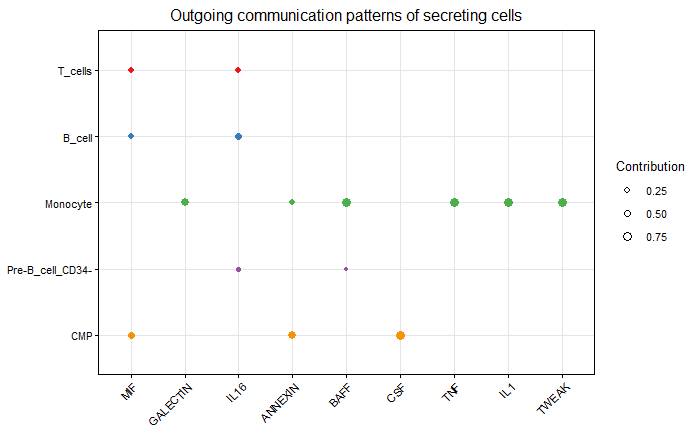
可以用桑基图和气泡图进行可视化,更直观的看到他们直接的联系。
1 | |
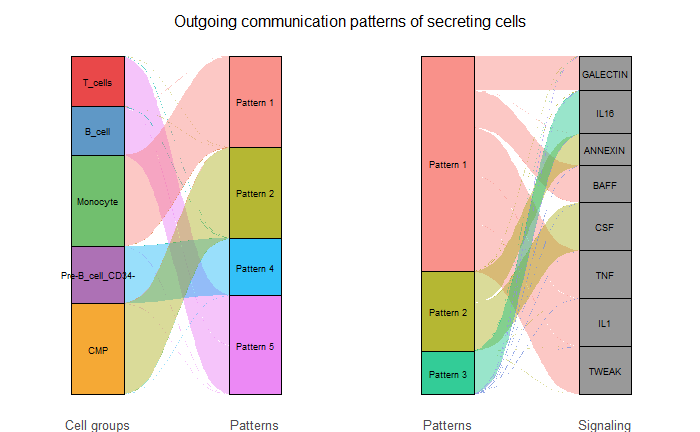
我们这里设置pattern 等于outgoing,用于显示输出模式,也就是展示作为信号发送器的细胞如何相互协调,以及它们如何与某些信号通路协调以输出信号。selectK(cellchat, pattern = “outgoing”)
如果想看输入模式,也就是作为信号接收器的细胞如何相互协调,以及它们如何与某些信号通路协调以响应输入信号,只需要把参数patten 改为 =“incoming”即可
1 | |
Tips:
正常的桑基图如下图,也就是左侧展示了cell group信息,每个细胞群属于哪个pattern,右侧灰色的地方是信号通路,展示了信号通路属于哪个pattern。(查看我们绘制的通讯模式热图可知, 由于数据集没有识别到在pattern 4和pattern 5 中输出模式下贡献度明显升高的信号通路,因此pattern = 3可能是更好的选择。Cophenetic和Silhouette只是辅助我们进行parttern数量的选择)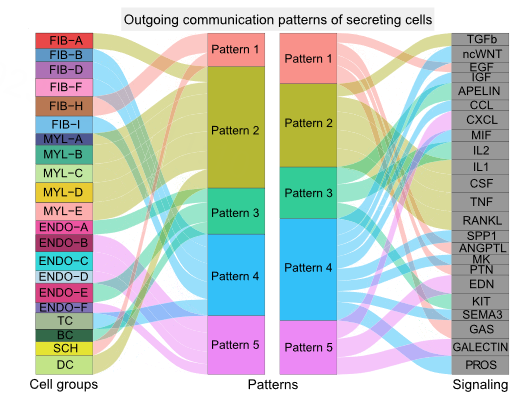
基因表达情况分析
- 信号通路相关基因的表达模式
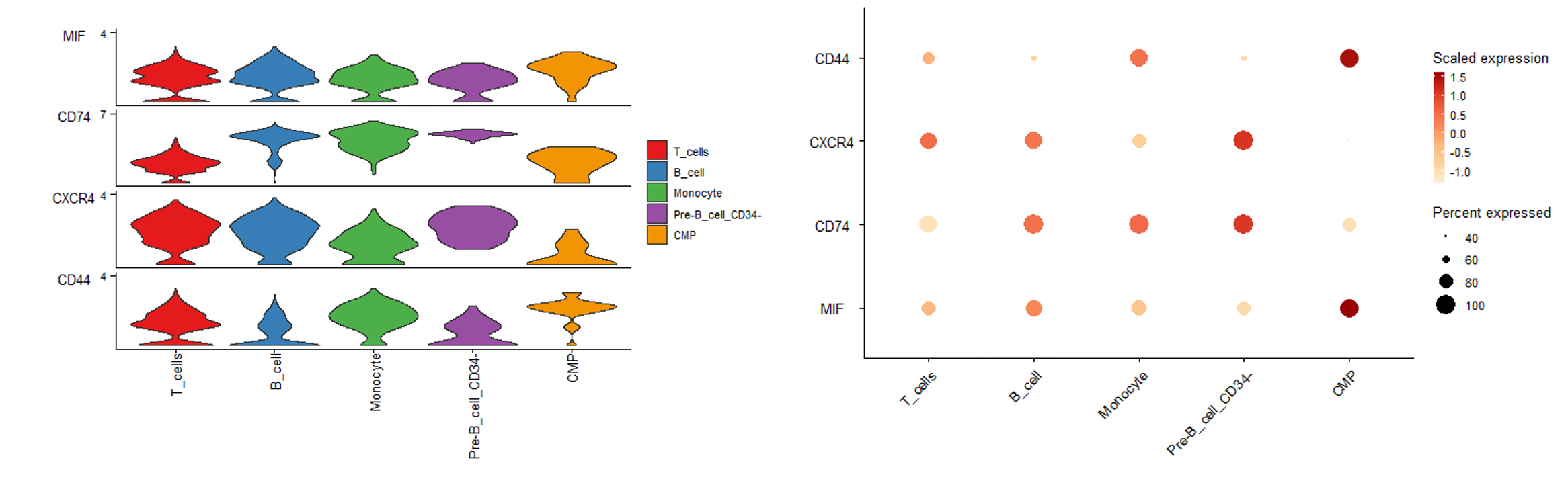
探索了信号通路之后,我们可以再去细致的挖掘,信号通路相关的基因的表达模式,可以使用小提琴图和气泡图可视化,
仍然以MIF信号通路为例,使用小提琴和气泡图进行该通路下通路相关基因的表达情况。
1 | |
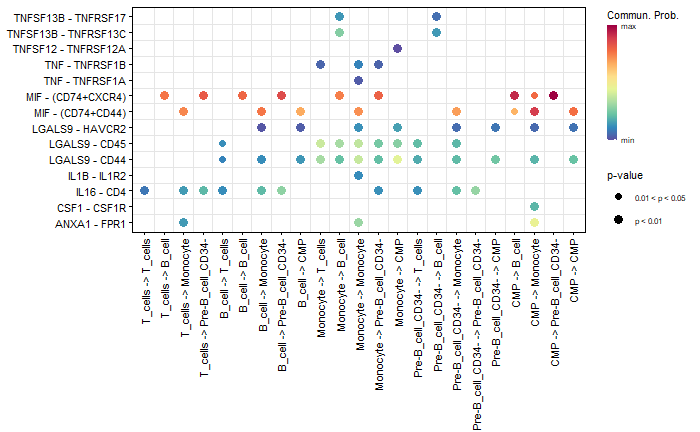
- 整体气泡图
我们也可以对由多个受配体或信号通路介导的细胞间通讯整体进行可视化,气泡图绘制了(全部配体-受体及相关信号通路)
1 | |
- 整体和弦图
Tips:Chord diagram的两种模式
netVisual_chord_cell可视化不同细胞群之间的细胞-细胞通信(其中弦图中的每个扇区是一个细胞群).
netVisual_chord_gene可视化由多个配体-受体或信号通路介导的细胞-细胞通信(其中弦图中的每个扇区是一个配体、受体或信号通路)
1 | |
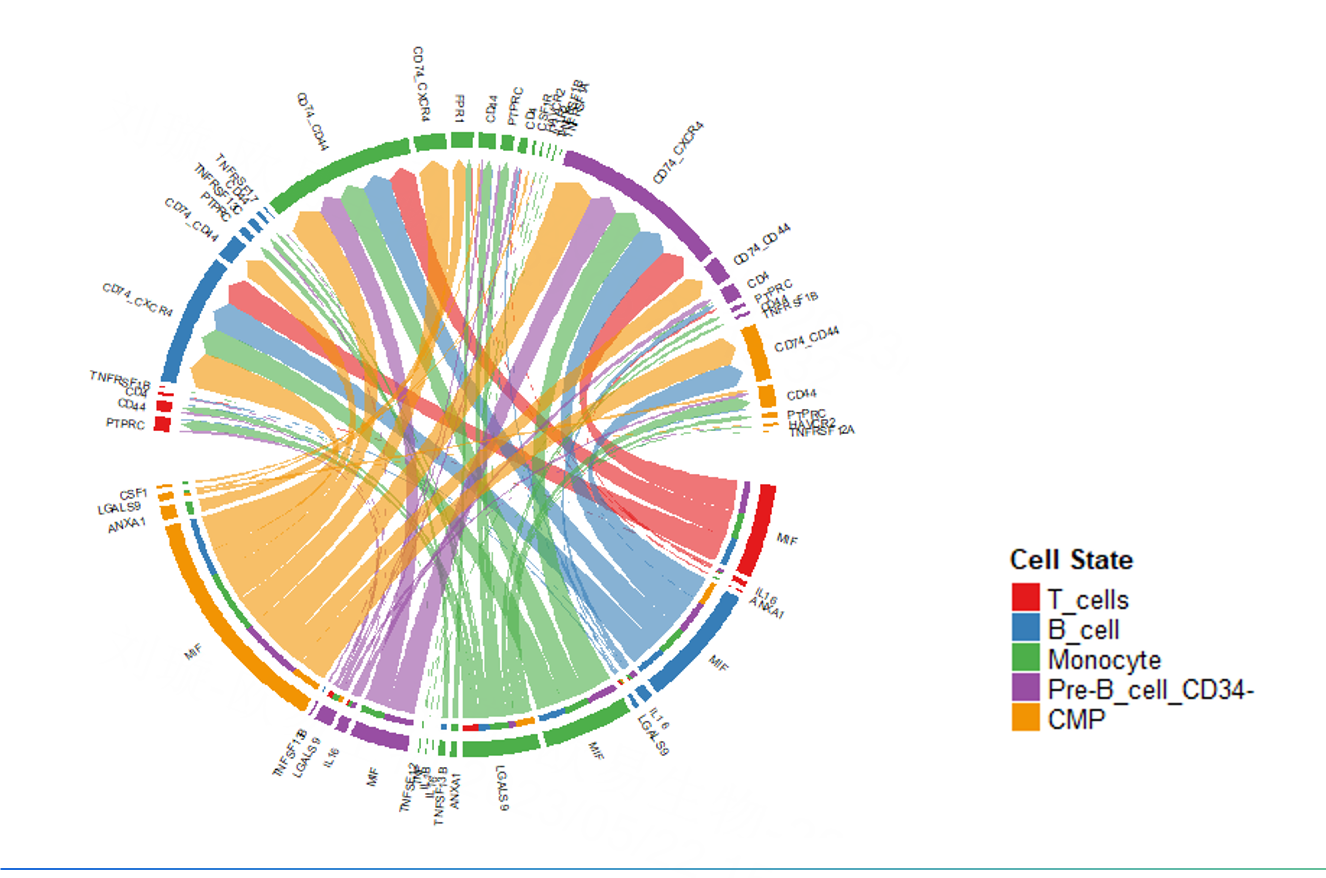
个性化绘图
个性化绘制指定受配体或信号通路介导的细胞间通讯,比如我们将发出信号的细胞类型局限为T细胞,接收信号的细胞类型设置为monocyte, B细胞和pre B。
1 | |
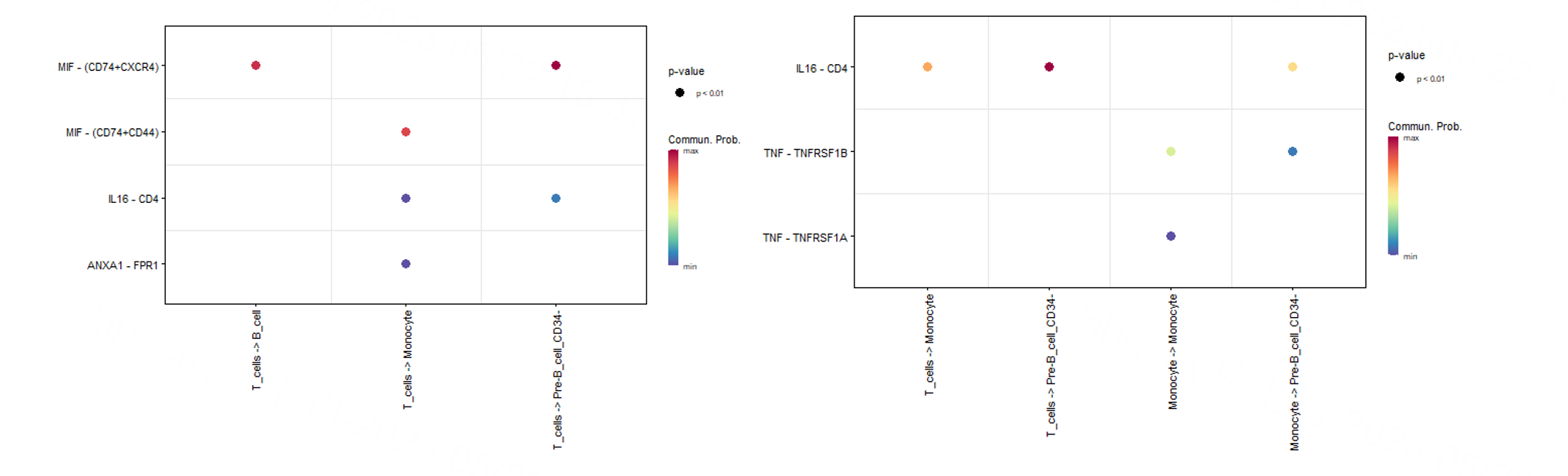
多组比较
由于我们本次的数据集细胞数量较少,且细胞在组间分布不均匀,后续就不进行个性化展示。
多组比较和之前的思路相似,先按组别进行拆分,然后分别对各组cellchat进行细胞通讯分析即可。使用netVisual_diffInteraction计算两组之间通讯概率的差值,并进行展示。
1 | |