Monocle2
Monocle2的学习笔记及代码分享
背景介绍
在研究胚胎发育,细胞转化以及干细胞在再生和疾病中的功能等领域,通过谱系追踪来研究单个细胞及其后代的命运是常见的研究方法。传统的谱系追踪涉及用染料显微注射到单个细胞或细胞群中以观察细胞迁移,或用利用Cre-LoxP 位点特异性重组系统对细胞命运进行追踪展示等基于成像的方法(Imageing-gased approaches)。
在NGS测序出现后,使用计算手段来追踪或重建细胞谱系对谱系追踪领域影响巨大。基于scRNA-seq,还可以在进行建立细胞谱系的同时分析基因表达模式。
由于细胞分化或细胞成熟等细胞进程在生物进程中并不总是以一种分散的状态,比如clustering,进行的,而更多是一种动态的,连续的发育状态。因此,很多计算方法致力于将细胞投射到一个连续路径上(Pseudotime)来展示细胞的发育轨迹。
下图为拟时序分析的简单示例图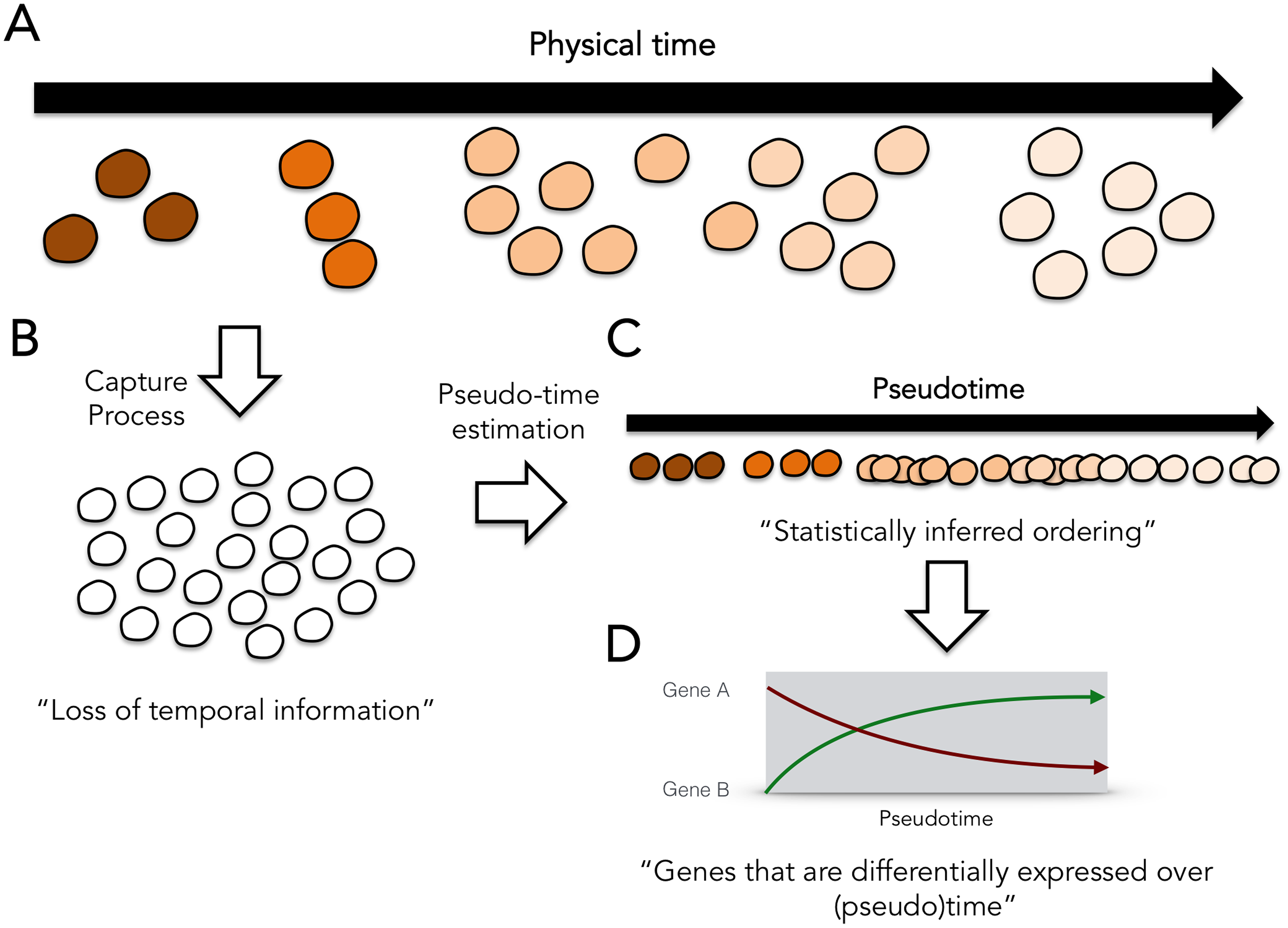
Monocle介绍
软件目的:
1: 对从细胞身份转化的过程进行建模(从一个clusters进展到另一个cluster),研究分化过程。
2: 可以使用轨迹之间的差异来识别与轨迹转变相关的基因
注意:推断的轨迹过程有时不一定反应真实的生物学过程,需要结合其他生物学先验知识和证据来对轨迹进行解释。
研发历史
2014年 Nature Biotechnology
monocle的概念版本发表在2014年,这个时间点,基于微流控的高通量的单细胞测序技术,比如droplet-based的scRNA-seq技术都还没有发表,商业化的10X单细胞测序也还未出现,此时的单细胞测序还是通过显微成像或者流式细胞技术进行细胞水平的研究。因此这个版本的Monocle针对的其实是低细胞数的轨迹(trajectory)分析。虽然和现在的10X单细胞测序动辄能测上万细胞的通量相比,当时用作分析的细胞数也就是几百个,相比之下低了太多,但就算如此,bulk RNA seq转录组测序的差异表达基因算法仍然不适用与单细胞测序。因为Bulk RNA-seq测序数据的差异分析通常采用基于平均值或方差的假设检验方法,无法捕捉到单个细胞水平的表达动态。
Monocle1,Monocle的初始版本,是第一个发布的单细胞RNA测序数据分析软件包之一,它认为就算单细胞测序是在同一时间捕获到的细胞群体,它也可能包括许多不同的中间分化状态,传统转录组那种只考虑其平均值的算法会掩盖在单个细胞中发生的趋势。

Monocle基于一个基本假设:scRNA - Seq中的所有细胞存在潜在的时间顺序,由于分化不同步使得细胞的基因表达水平存在很强的差异性,那就可以根据这个潜在的动态过程对细胞进行排序。Monocle的算法源自先前的基于bulk RNASeq重构样本间的时间顺序的算法,但将其扩展到单细胞层面,并允许单个祖细胞类型存在多个细胞命运。
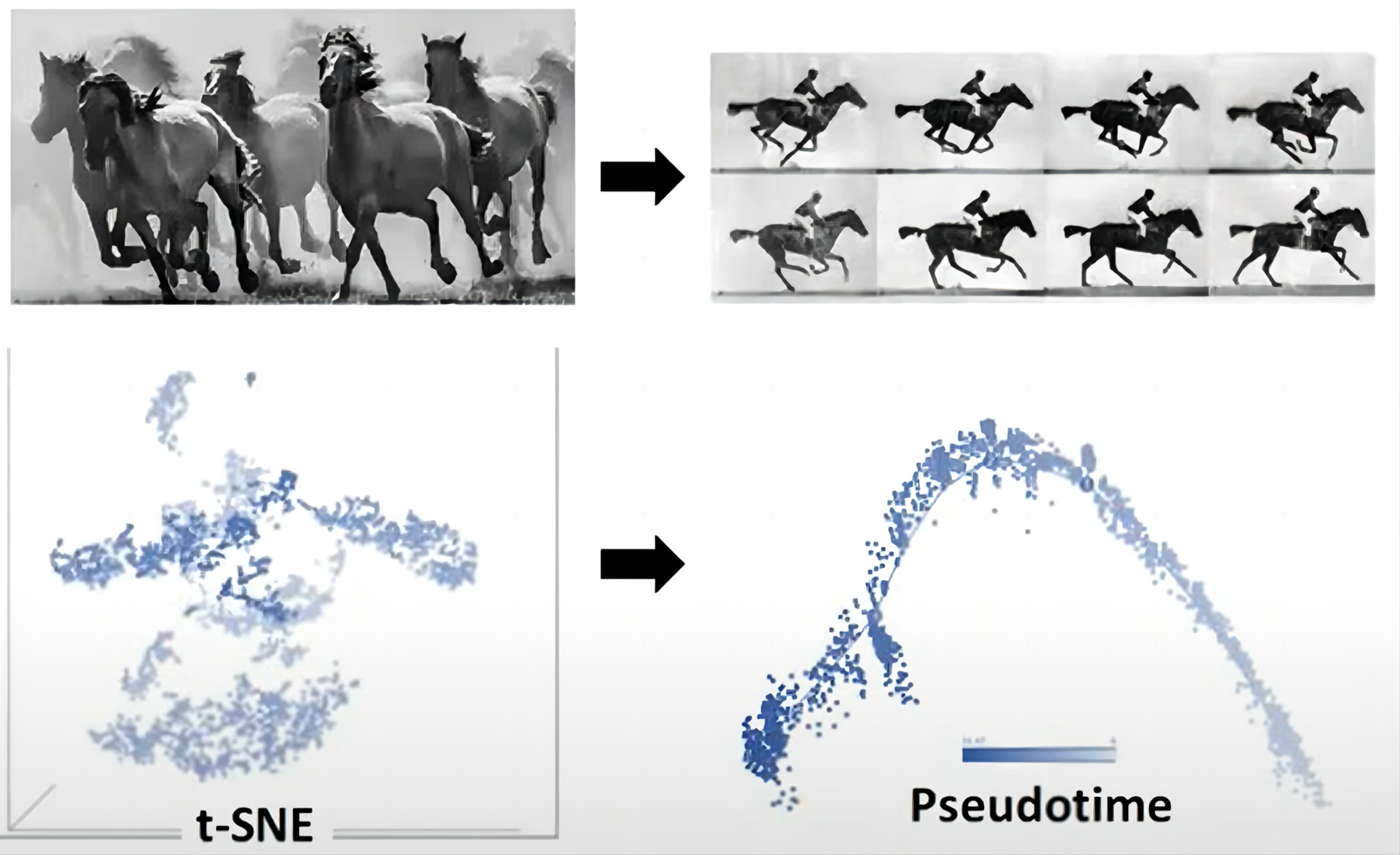
Monocle引入了一个概念: “pseudotime”, 即通过生物过程进展的定量测量,在’pseudotime拟时间’中排序单细胞表达谱。或者说是细胞在发育过程中的时间顺序。monocle1可以根据基因表达数据生成拓扑图,确定基因表达水平的变化趋势,并可视化地展示出细胞发育轨迹,从而更容易理解和解释单细胞RNA-seq数据的结果。
热图展示了Monocle找到的差异基因(行),细胞(列)按拟时间顺序排列。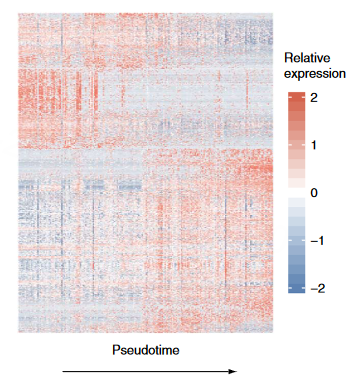
工作原理图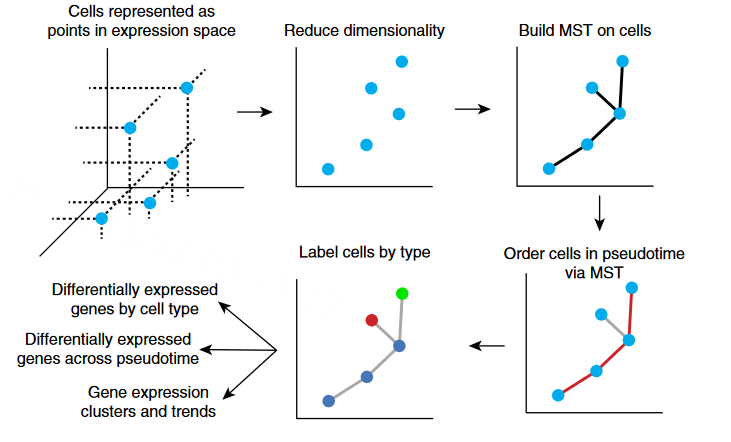
2017年 Nature Methods (1月)
随着测序技术的发展,高通量的单细胞测序技术产生了,单细胞测序可以检测到更多的细胞和更多的基因表达水平。为了适应这种变化,monocle2,在2017年被发表在nature methods上。这个版本的monocle对之前算法进行了进一步优化,增加了用于分支分析的BEAM算法。
Branched expression analysis modeling (BEAM)
为了以稳健的方式从学习到的轨迹中检测细胞命运依赖基因,作者开发了BEAM,一种广义线性模型,用于找到分支之间不同的所有基因,(其实就是一种差异分析方法)。什么是分支呢,就是当发育过程中,细胞执行不同的基因表达程序时,这个时候就是细胞做出命运抉择的时刻。当细胞做出命运选择时,分支就会出现在轨迹中: 一个发育谱系沿着一条路径前进,而另一个谱系产生第二条路径。BEAM算法就可以用来识别在每个谱系分岔过程中表现出显著差异的基因,帮助确定在复杂的生物学过程中细胞命运决定的时刻。
BEAM方法接受一个已通过orderCells函数排序的CellDataSet以及发育轨迹中分支点的名称作为输入,返回每个基因的显着性得分表。得分显着的基因在其表达中被认为是分支依赖性的。
下图展示了’pseudotime拟时间”排序后的细胞轨迹,可以看到上面有黑色的圆圈,是分支点1, branch_point 1,也就是细胞分化细胞命运决定的时刻。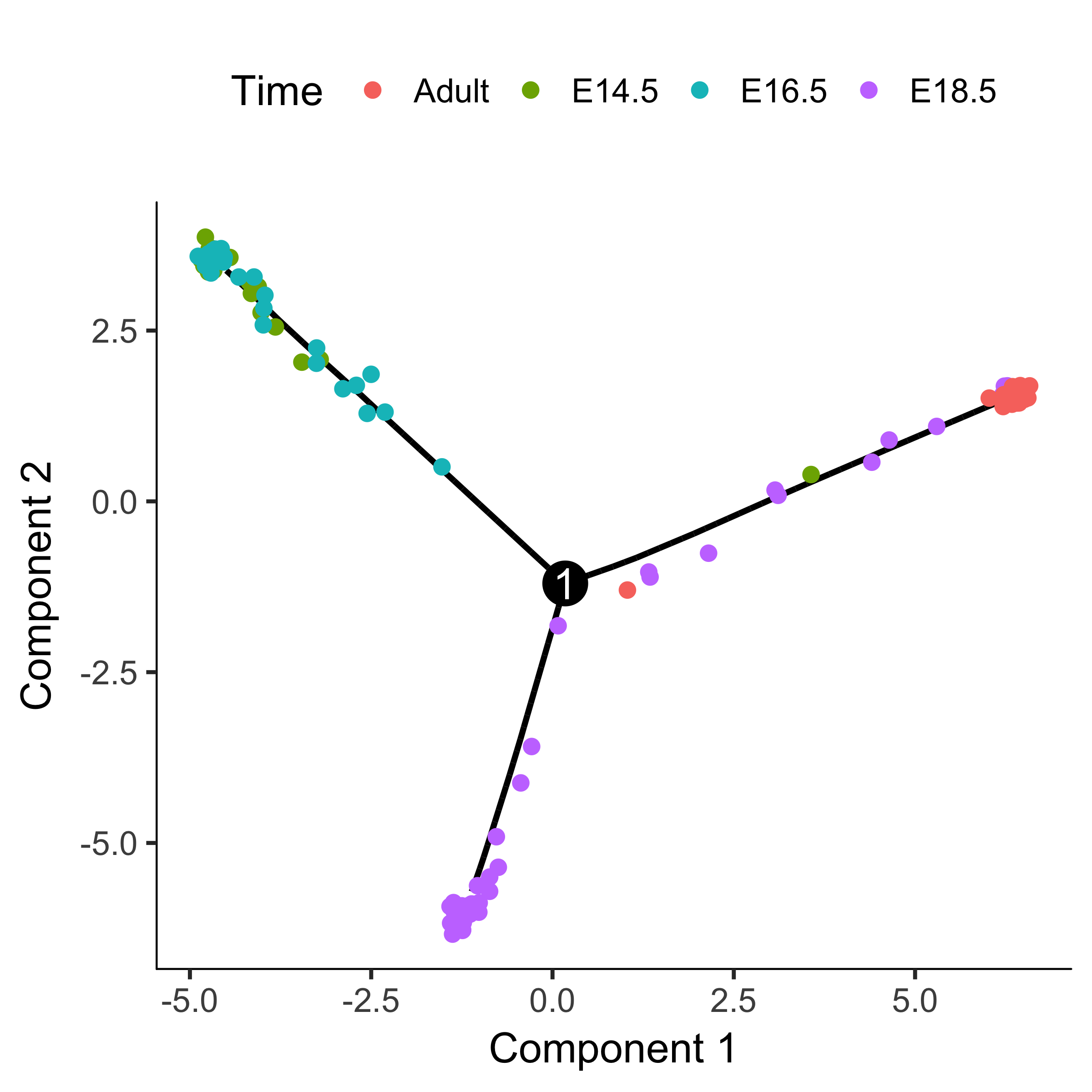
图中简单展示了Beam算法的原理,它使用广义线性模型对已知分支值的数据进行回归(alternative model),为每个分支拟合单独的曲线。它还通过对所有数据拟合一条曲线来执行分支指派未知的回归(零模型),然后通过似然比检验比较这些模型。 判断当细胞从树的左上角(E14.5)的早期发育阶段通过分支时,哪些基因发生了变化?哪些基因在分支之间差异表达?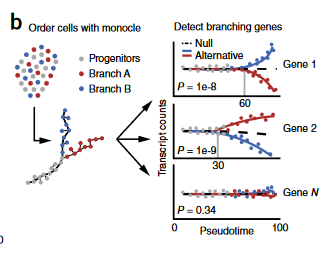
可以看到分化的关键基因(marker基因)Pdpn and Sftpb在明显属于随不同branch变化的基因, 两个分支之间呈现出了显著差异的趋势,但housekeeping genes (Hprt and Pgk1)并未有明显差异。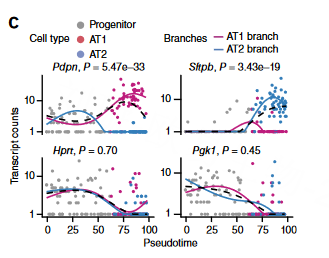
2017年 Nature Methods (8月)
这篇文献主要讲述了Monocle2的工作流程,介绍了如何将反向图嵌入引入细胞轨迹分析,并改为使用DDRTree的算法做降维。monocle2的稳定版本从此开发出来,它也成为了单细胞领域目前应用最广泛的轨迹推断方法。
monocle2的流程图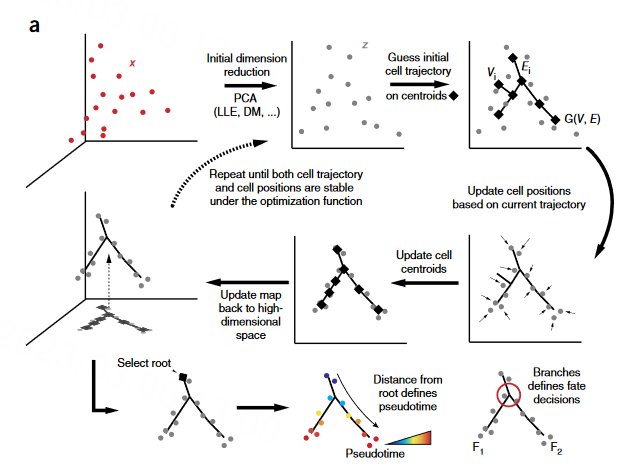
关键步骤:
- 选择定义过程的基因: 在单细胞测序中,会出现很多低水平表达的基因,造成一个噪声的影响,因此我们在分析的时候不能选择使用全部的基因分析。推断单细胞的轨迹是一个机器学习的问题,轨迹推断的第一步就是选择monocle使用哪些基因作为轨迹推断的关键基因。也就是机器学习中的特征选择问题。
Monocle2首先使用了”dpFeature”方法确认与生物过程相关的基因(这些基因在下游分析时被称为ordering gene),注意一下,这里与BEAM这个寻找分叉点相关基因的算法不同,dpFeatures找到的是全局的拟时相关基因,也就是拟时起点和终点相关的基因。
每个细胞被表示为高维空间(x)中的一个点,其中每个维度对应一个基因的表达水平。数据从高维空间(X)被投影到低维空间(z)上,使用k - means聚类算法找到这些细胞的分群中心,然后通过反向图嵌入(默认是DDRTree)的流形学习算法,找到高维基因表达空间与低维空间的映射关系并构建最小生成树,将细胞投影到生成树上,并将找到的中心进行连接,构建初步的树形结构。
基于新的细胞位置,更新细胞的分群中心和轨迹结构,将二维空间中的树(二维轨迹),重新投射到高维空间。迭代此过程,直至收敛(即获得稳定的树形结构,细胞轨迹可以反应原始数据的特征)。
这里提到了两个关键词,dpFeature, 用于确认与生物过程相关的基因,也就是ordering gene。以及DDRTree,在monocle的reduceDimension函数中默认使用,用来推断基因表达动态变化,这两个算法是monocle2的主打点。
其实目前monocle已经更新到3代了,但考虑到目前发表的文章还是使用monocle2的比较多,而且monocle3的下游可视化方式还有待开发,我们仍然使用monocle2进行讲解。
应用场景
挖掘细胞分化的关键基因:
在分化或发育研究中,可通过拟时序分析关键的分化节点,挖掘细胞不同分化路径命运选择的推动基因揭示细胞发育轨迹:
对于未知的细胞间或细胞亚群,通过拟时序分析揭示细胞分化轨迹,推测干细胞在发育过程的分化轨迹或某类细胞的分化来源揭示细胞动态变化:
在肿瘤或免疫研究中,通过拟时序分析追踪细胞的动态变化,推断细胞亚型的演变轨迹或细胞凋亡路径
代码关键步骤讲解
CellDataSet对象构建与预处理
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37#可选方案: 从表达量矩阵和细胞、基因的表型文件构建CellDataSet对象
#1. expr_matrix
#行为基因,列为细胞的表达量矩阵
#2. phenoData
#对应的细胞的标签信息(barcodes)
#3. featureData
#对应的基因的标签信息,其中必须包含“gene_short_name”列
##pd, phenoData
pd <- new("AnnotatedDataFrame", data = seurat_ob@meta.data)
##fd, featureData
gene_annotation=data.frame(gene_short_name = rownames(seurat_ob@assays$RNA@counts),
stringsAsFactors=F)
rownames(gene_annotation)<-gene_annotation$gene_short_name
fd <- new("AnnotatedDataFrame", data = gene_annotation)
gbm_cds <- newCellDataSet(GetAssayData(seurat_ob,slot="counts",assay="RNA"),
phenoData = pd,
featureData = fd,
expressionFamily=negbinomial.size())
#推荐方案
#从Seurat聚类结果构建CellDataSet对象
cds <- as.CellDataSet(seurat_ob)
#size facotr帮助我们标准化细胞之间的mRNA的差异。
#离散度值可以帮助我们进行后续的差异分析。
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
#过滤低质量细胞(保留至少在1个细胞中counts大于1的基因)
cds = detectGenes(cds,min_expr = 1)
#保留至少在10个细胞中表达的基因
expressed_genes = row.names(subset(fData(cds),num_cells_expressed>10))寻找生物学过程相关的基因
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24#推荐方案: 使用monocle的dpFeature方法
#differentialGeneTest函数即为monocle的dpFeature方法,用于确认与生物过程相关的基因(即与clusters分群相关的差异基因)
#The group design to find ordering genes, 这里使用的是seurat的clusters
clustering_DEGs = differentialGeneTest(cds[expressed_genes,],fullModelFormulaStr ="~clusters",cores = 4)
featureData(cds)@data[rownames(clustering_DEGs),"pval"]=clustering_DEGs$pval
featureData(cds)@data[rownames(clustering_DEGs),"qval"]=clustering_DEGs$qval
#根据qval筛选ordering gene
ordering_genes <- row.names (subset(clustering_DEGs, qval < 0.01))
#一旦我们有用于Ordering的基因ID列表,我们就需要将它们设置在cds对象中
gbm_cds = setOrderingFilter(cds,ordering_genes = ordering_genes)
#可以使用fData(gbm_cds)查看,基因矩阵中增加了一列use_for_ordering列
plot_ordering_genes(gbm_cds)
#可选方案:选择方差最大的基因
disp_table <- dispersionTable(cds)
ordering_genes <- subset(disp_table,
mean_expression >= 0.5 &
dispersion_empirical >= 1 * dispersion_fit)$gene_id
#将ordering gene设置在cds对象
gbm_cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(gbm_cds)构建生成树
1
2
3
4#the dependent package irlba should not be less than 2.3.2, or the following step will fail
#if the size of cells is big.
gbm_cds = reduceDimension(gbm_cds,max_components = 2,verbose = T)
gbm_cds = orderCells(gbm_cds,reverse = F)可视化结果展示
轨迹图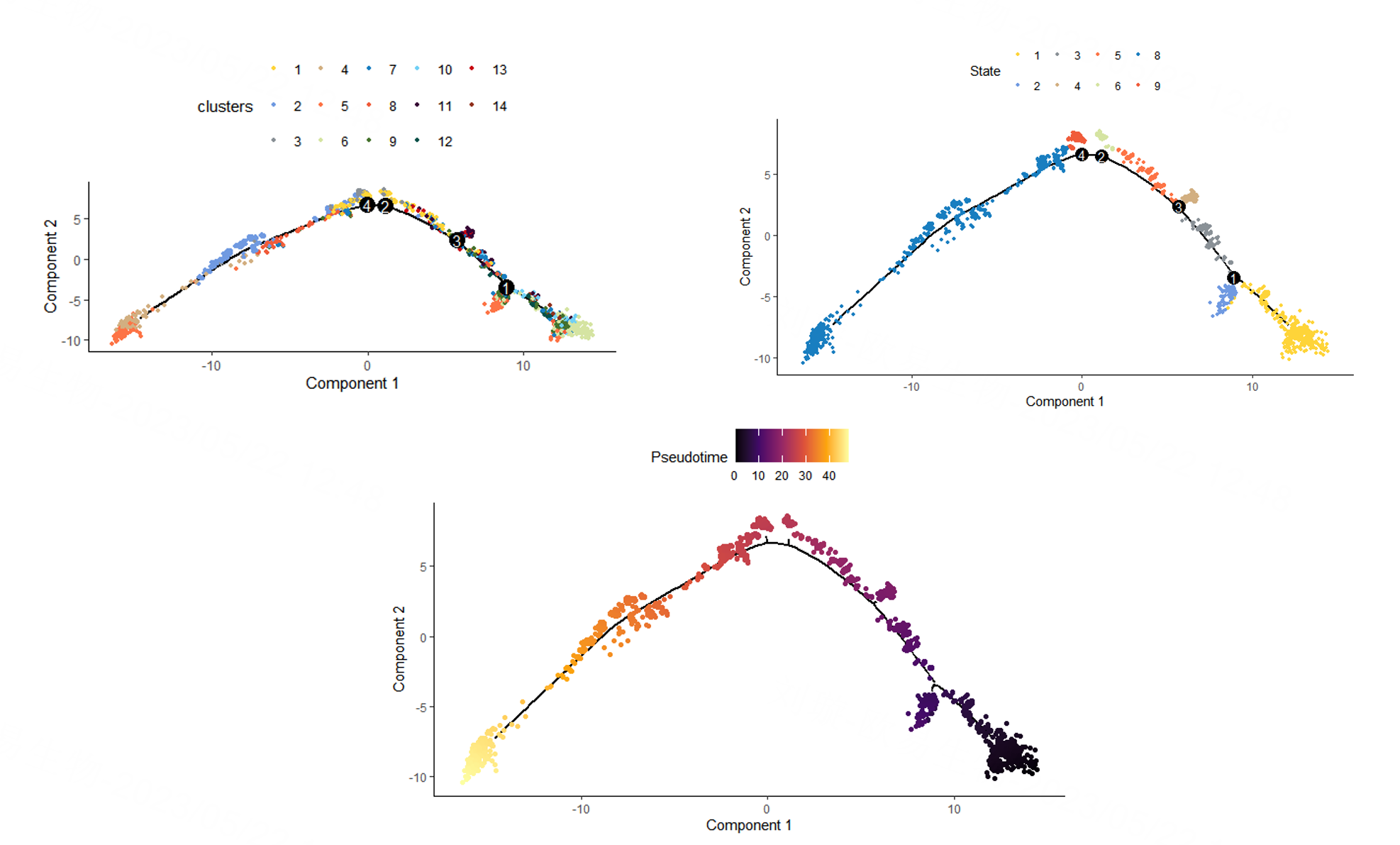
基因表达量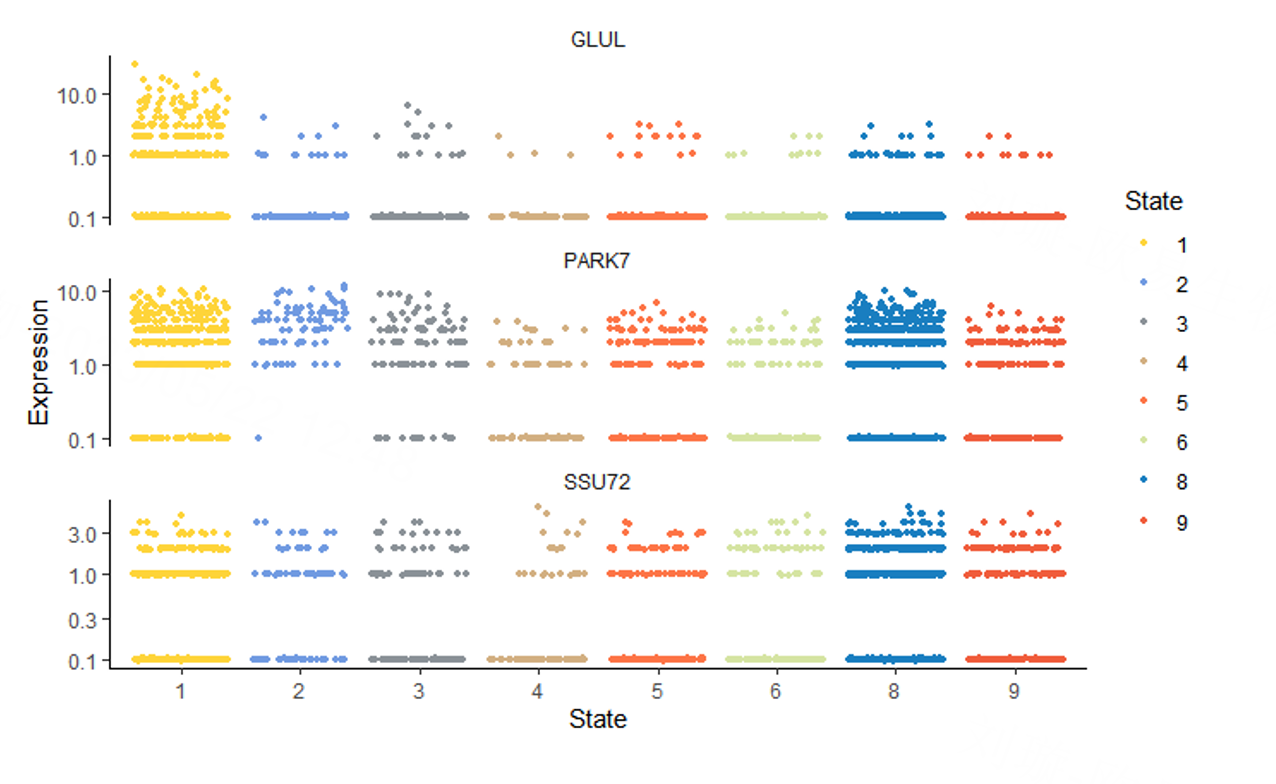
动力学趋势图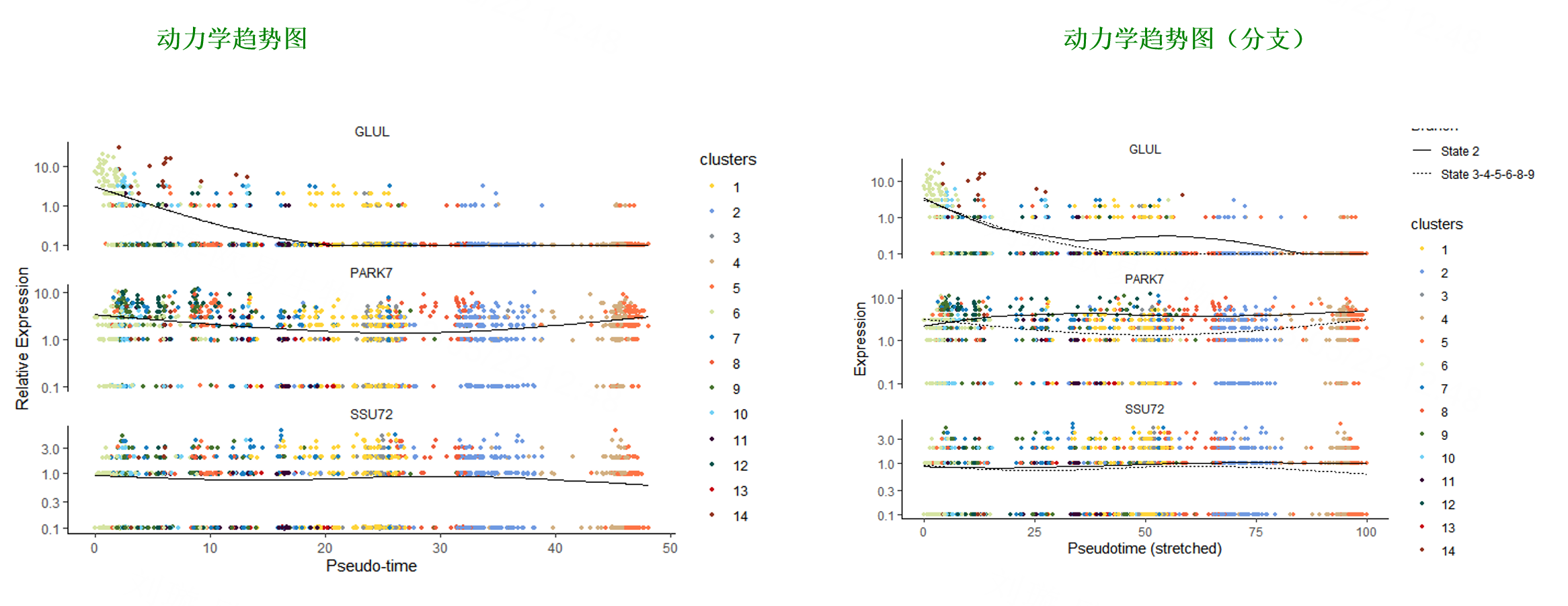
热图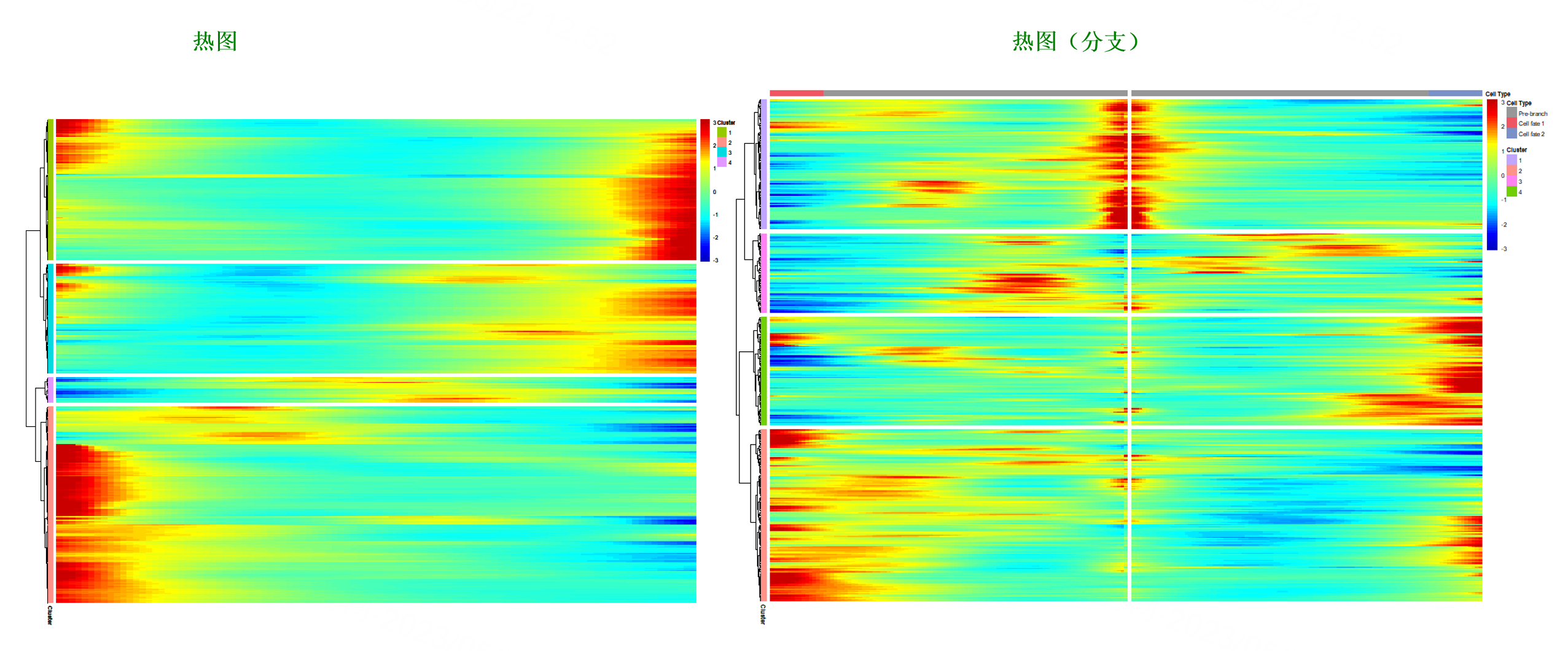
进阶练习:将基因表达量映射到拟时序轨迹上
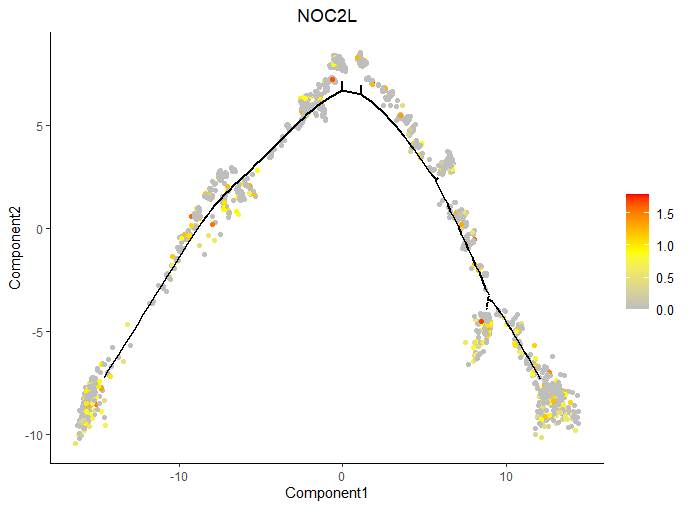
1
2
3
4
5
6
7
8
9
10
11Dims = as.data.frame(t(monocle::reducedDimS(gbm_cds))) %>% rownames_to_column("Barcodes")
colnames(Dims)[2] = "Component1"
colnames(Dims)[3] = "Component2"
i= to_be_tested[1]
data = FetchData(object = seurat_ob, vars = c("clusters",as.character(i))) %>% rownames_to_column("Barcodes")
data_df <- merge(data,Dims, by="Barcodes")
g <- ggplot(data=data_df, aes(x=Component1, y=Component2)) + geom_point(aes(color=data_df[[i]]), size=1.5, na.rm = TRUE)+theme_classic()+ scale_color_gradientn(colours = c("grey", "yellow", "red"))+theme(legend.text = element_text(size = 10),legend.title = element_blank(),plot.title = element_text(hjust = 0.5))+labs(title = i)
其他拟时序软件介绍
RNA velocity (RNA速率)
分子生物学上我们都知道,刚转录出的mRNA包含外显子和内含子,经过剪接切除内含子后,得到用于编码蛋白的成熟mRNA,最后经过降解消除。在单细胞测序中,可以很容易观测到剪接的发生,例如跨外显子/内含子的reads可表示未剪接的pre-mRNA,跨外显子/外显子的reads可表示剪接的成熟mRNA,通过Cellranger软件,也就是在下机数据与基因组比对的初始处理步骤中,可针对所有基因的每个细胞产生剪接和非剪接RNA数量进行计数。如果转录速率恒定,那RNA的丰度(也就是RNA velocity)只由未成熟转向成熟,以及成熟后的降解两个因素所决定。通过计算剪接和未剪接的mRNA分子数,我们就可以间接的估算基因的转录速率。对于某个基因来说,如果它新生的RNA”耗尽”了,就表示这个基因的表达被下调,而未成熟mRNA的积累表示这个基因的表达被上调。
如图所示,RNA velocity的基本原理:先是计算所有基因的每个细胞的剪接和未剪接计数,然后推断合适的转录动力学模型。通过计算未剪接转录本和剪接转录本之间的比率可以推断细胞命运的状态(过渡与稳定)和方向性(轨迹)。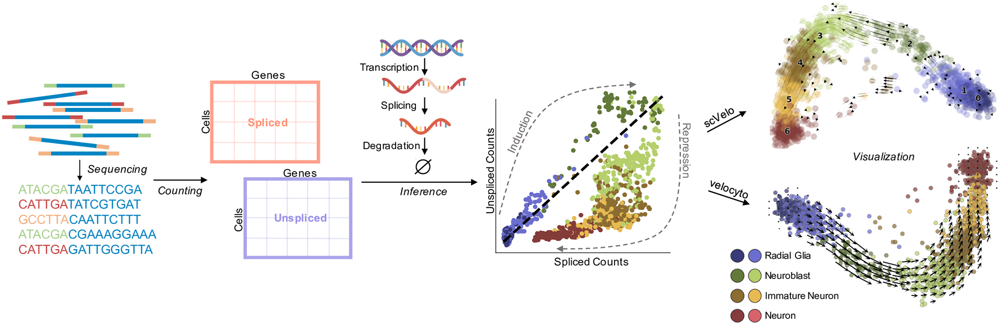
使用RNA速率分析,就可以计算细胞内mRNA剪接前后比例来估计RNA丰度随时间的变化,推断转录事件的时间顺序,估算RNA相关的转录剪切降解速率,在无需先验知识的情况下,推断细胞的下一个可能的分化方向。
RNA velocity可以揭示单细胞基因表达在时间尺度上的动态变化,这种动态变化与人类和其他哺乳动物的发育、再生和反应过程相匹配。它是一种局部速度向量,可用于模型定型、命运选择和体内转录的精确动力学研究,能够详细研究复杂组织和器官的动态过程,并将极大地促进人类胚胎的谱系分析。
RNA velocity的方法的具体实现通常是通过velocyto和scVelo,由于scVelo软件包的维护做的比较好,并且引入了更优化的模型,基于似然的动力学模型,所以scVelo会更常用一些。由于RNA velocity的环境配置比较复杂,仅做原理讲解,不做实操展示。
应用场景:
动力学研究:
模型定型、命运选择和体内转录的精确动力学研究揭示细胞动态变化:
能够详细研究复杂组织和器官的动态过程,谱系分析:
人类胚胎的谱系分析
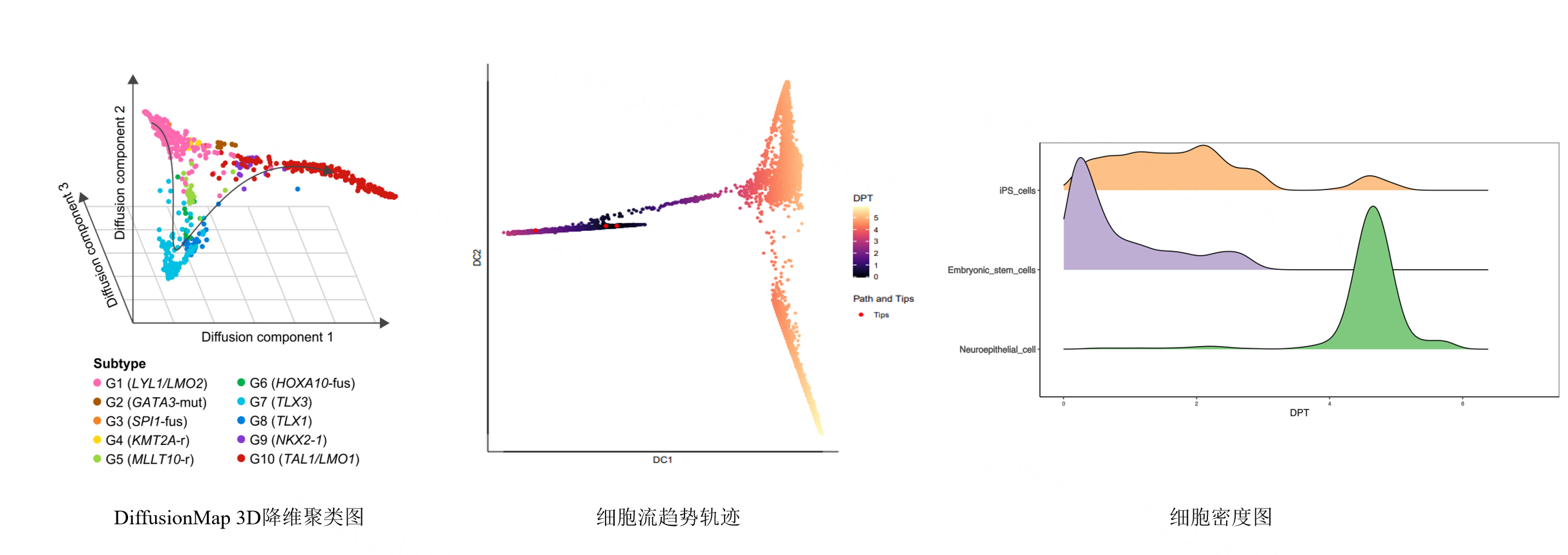
Diffusion map
回忆一下之前谈到的monocle的原理,monocle2依赖differentialGeneTest方法,需要现有一个分组的信息才能进行轨迹构建。但diffusion map与monocle2不同,diffusion map不需要clusters等信息,不需要根据特定的属性对细胞进行分组聚类。因此,Diffusion map可以保留连续的分化轨迹。
Diffusion Map通过马尔科夫链的概率策略来对单细胞数据进行建模,判断细胞转移的方向,将细胞间的空间结构降维并映射为连续性结构。算法层面相对简单,比较适用于研究样本集中单个细胞的分化或者谱系起源,或者针对从不同时间点取到的相同类型的细胞进行时序分析。对于复杂的数据集或者可能存在分支的数据集,不建议使用。
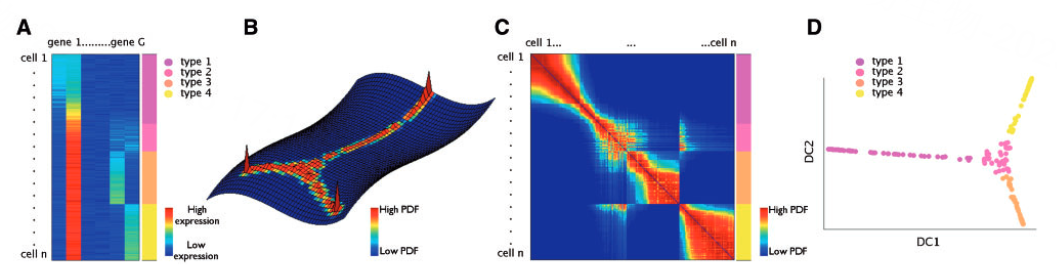
基本原理:
- 输入细胞*基因的(n x G)矩阵,和monocle类似,变到高维空间
- 通过高斯核函数,高斯核函数定义成对数据点之间的相似度矩阵。从G维基因空间中找到由较高概率密度的连续路径所组成的扩散路径。
- 根据细胞对之间的转移概率,计算 n × n 的细胞间马尔可夫转移概率矩阵 。(如果对数据进行随机游走,则走到附近的数据点比走到远处的另一个数据点更有可能,也就是概率更高)
- 对马尔可夫转移概率矩阵进行特征值分解, 获得特征向量(一般取前三维度),获得细胞在不同维度的坐标。
通过扩散映射将空间距离转换为状态转移的概率,对不同分化路径中的细胞进行降维和排序,确定分化细胞的随机转移方向,进而预测细胞的发育轨迹并检测稀有种群。可以与RNA velocity共同进行分析,做相互验证。
基于Diffusion maps算法开发的destiny软件发表在bioinformatics,因为destiny包可以兼容seurat包且与其他方法相比,diffusion map算法对噪声扰动具有鲁棒性并且计算成本低(内存消耗远小于monocle),可以将细胞间非线性的空间结构降维并映射维连续性结构。相关文献已经被引用超过1000次,也是一个稳定的分析方法了。
结果展示: